Chromatin State Discovery¶
usage: chromHMM.py [-h] [-j JID] [--pipeline_type PIPELINE_TYPE]
[-se SE_FASTQ_LIST] [-tf TF_FASTQ_LIST]
[-his HIS_FASTQ_LIST] [-d_tf DESIGN_MATRIX_TF]
[-d_his DESIGN_MATRIX_HIS] [-d1 DESIGN_MATRIX_1]
[-d2 DESIGN_MATRIX_2] [--LearnModel_addon LEARNMODEL_ADDON]
[-pe PE_FASTQ_LIST] [-c CELL_LINE] [--from_binBam]
[-m MEMORY] [-bin CHROMHMM_JAR]
[--known_association KNOWN_ASSOCIATION]
[--chromatin_state_info CHROMATIN_STATE_INFO]
[-n NUMBER_STATES] [--d1_bin_bam_addon D1_BIN_BAM_ADDON]
[--d2_bin_bam_addon D2_BIN_BAM_ADDON]
[--adjust_TF_pvalue ADJUST_TF_PVALUE]
[--adjust_histone_pvalue ADJUST_HISTONE_PVALUE] [-g GENOME]
[-i INDEX_FILE] [-s CHROM_SIZE]
perform chromatin state discovery using chromHMM v1.18, bin size is fixed to
be 200bp.
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
chromHMM_yli11_2019-10-13)
--pipeline_type PIPELINE_TYPE
Not for end-user. (default: chromHMM)
Input Files:
-se SE_FASTQ_LIST, --SE_fastq_list SE_FASTQ_LIST
A tsv file containing 2 columns, R1.fastq.gz & UID
(default: None)
-tf TF_FASTQ_LIST, --TF_fastq_list TF_FASTQ_LIST
A tsv file containing 2 columns, R1.fastq.gz & UID
(default: None)
-his HIS_FASTQ_LIST, --His_fastq_list HIS_FASTQ_LIST
A tsv file containing 2 columns, R1.fastq.gz & UID
(default: None)
-d_tf DESIGN_MATRIX_TF, --design_matrix_TF DESIGN_MATRIX_TF
Similar to peakcall.tsv, this is a tsv file containing
3 columns: treatment_UID & control_UID & label. Label
has to be meaningful labels, such as H3K4me3. Case
insensitive. (default: None)
-d_his DESIGN_MATRIX_HIS, --design_matrix_His DESIGN_MATRIX_HIS
Similar to peakcall.tsv, this is a tsv file containing
3 columns: treatment_UID & control_UID & label. Label
has to be meaningful labels, such as H3K4me3. Case
insensitive. (default: None)
-d1 DESIGN_MATRIX_1, --design_matrix_1 DESIGN_MATRIX_1
Similar to peakcall.tsv, this is a tsv file containing
3 columns: treatment_UID & control_UID & label. Label
has to be meaningful labels, such as H3K4me3. Case
insensitive. (default: None)
-d2 DESIGN_MATRIX_2, --design_matrix_2 DESIGN_MATRIX_2
A tsv file contatinig 2 columns: UID & label.
(default: None)
--LearnModel_addon LEARNMODEL_ADDON
chromHMM learmodel additional parameters e.g. -u coord
dir (default: )
-pe PE_FASTQ_LIST, --PE_fastq_list PE_FASTQ_LIST
A tsv file containing 3 columns, R1.fastq.gz &
R2.fastq.gz & UID (default: None)
-c CELL_LINE, --cell_line CELL_LINE
input cell line, just a name, not important (default:
myCellLine)
--from_binBam Resume analysis from chromHMM binBam step (default:
False)
-m MEMORY, --memory MEMORY
memory requested (MB), if you have 16+ samples, for
example, 8 markers and 2 replicates per marker, use
200G, which is -m 200000 (default: 200000)
-bin CHROMHMM_JAR, --chromHMM_jar CHROMHMM_JAR
chromHMM bin location (default: /home/yli11/Programs/j
ar_tools/ChromHMM-1.18/ChromHMM.jar)
--known_association KNOWN_ASSOCIATION
chromHMM bin location (default: /home/yli11/HemTools/s
hare/misc/chromHMM_known_associations.tsv)
--chromatin_state_info CHROMATIN_STATE_INFO
chromHMM bin location (default: /home/yli11/HemTools/s
hare/misc/chromatin_state_info.tsv)
-n NUMBER_STATES, --number_states NUMBER_STATES
Number of chromHMM states to learn. Remember, chromHMM
uses binarized signals. If you have N markers, then
possibly you could explain up to 2^N states. In
practice, you should run this program several times,
each with different number of states. (default: 10)
--d1_bin_bam_addon D1_BIN_BAM_ADDON
This is an addon parameter for binarized bam with
design_matrix_1 as input (default: )
--d2_bin_bam_addon D2_BIN_BAM_ADDON
This is an addon parameter for binarized bam with
design_matrix_2 as input (default: )
--adjust_TF_pvalue ADJUST_TF_PVALUE
Most TFs, except for CTCF, tend to have much lower
number of 1s (default: 0.0005)
--adjust_histone_pvalue ADJUST_HISTONE_PVALUE
Some histone marks can have much higher number of 1s
after binarizeBam, increase the p-value cutoff can
descrease the number of 1s (default: 1e-05)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
-i INDEX_FILE, --index_file INDEX_FILE
BWA index file (default: /home/yli11/Data/Human/hg19/i
ndex/bwa_16a_index/hg19.fa)
-s CHROM_SIZE, --chrom_size CHROM_SIZE
chrome size (default: /home/yli11/Data/Human/hg19/anno
tations/hg19.chrom.sizes)
Summary¶
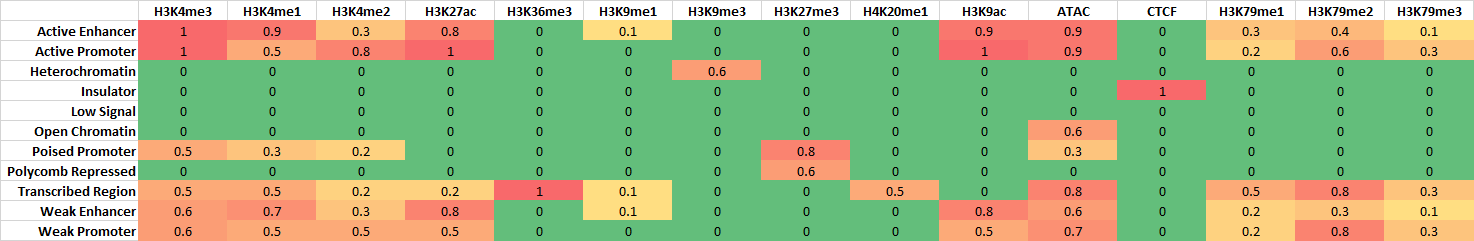
Perform chromatin state discovery given a list of fastq files. Single-end fastq is given using -se option. Paired-end fastq is given using -pe option. These input files are similar to fastq.tsv, as used in other HemTools programs. One can use either one or both input types. Design matrix is given as -d1 or -d2 options. -d1 input format is similar to peakcall.tsv, where the first two columns are UIDs (treatment vs control), and the third column is a label. This label has to be a meanfully label, such as H3K4me3 (Case insensitive). These labels are used to compare to known chromHMM annotations (see the second figure below). Still, chromatin state annotations are subjective, there’s no ground rules, this comparison is just to help you define the learned model.
Note
In theory, you can input any fastq data, such as RNA-seq, Hi-C, or TF CHIP-seq data. However, I haven’t seen papers using RNA-seq or Hi-C for chromatin state discovery. There are few papers using Pol-II, CTCF, or NANOG. You can definitely try everything.
Flowchart¶
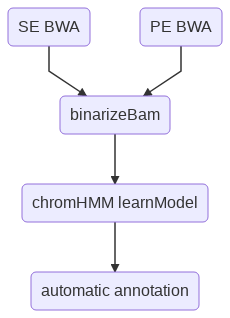
Chromatin states known associations¶
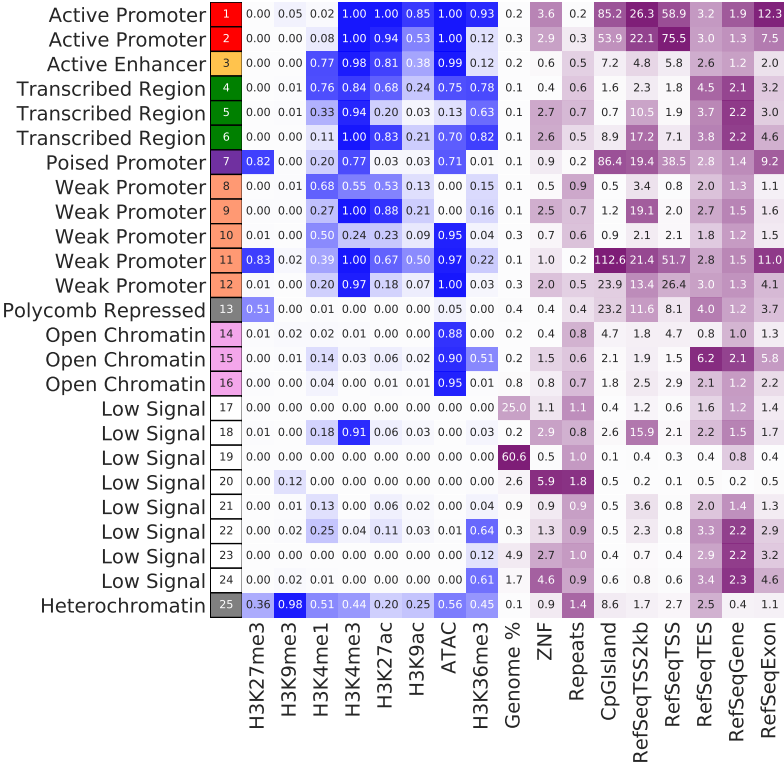
Example output¶
These chromatin state definitions were obtained by comparing with known associations. For exploratory usage, this is OK. For publication purpose, you will probably need to redefine some states.
Input¶
See Summary above for how these inputs are used.
-se: similar to fastq.tsv for single-end data
2 columns: file_location (with path if not in the current dir), UID.
See an example below. This file is named as se.list.
/path_to_file/1577764_ChIP_T_input_rep2_R1.fastq.gz input_rep2
/path_to_file/1577763_ChIP_T_input_rep1_R1.fastq.gz input_rep1
/path_to_file/1577762_ChIP_T_H3K36me3_rep2_R1.fastq.gz H3K36me3_rep2
/path_to_file/1577761_ChIP_T_H3K36me3_rep1_R1.fastq.gz H3K36me3_rep1
/path_to_file/1577760_ChIP_T_H3K27ac_rep2_R1.fastq.gz H3K27ac_rep2
/path_to_file/1577759_ChIP_T_H3K27ac_rep1_R1.fastq.gz H3K27ac_rep1
/path_to_file/1577758_ChIP_T_H3K27me3_rep2_R1.fastq.gz H3K27me3_rep2
/path_to_file/1577757_ChIP_T_H3K27me3_rep1_R1.fastq.gz H3K27me3_rep1
/path_to_file/1577756_ChIP_T_H3K9ac_rep2_R1.fastq.gz H3K9ac_rep2
/path_to_file/1577755_ChIP_T_H3K9ac_rep1_R1.fastq.gz H3K9ac_rep1
/path_to_file/1577754_ChIP_T_H3K9me3_rep2_R1.fastq.gz H3K9me3_rep2
/path_to_file/1577753_ChIP_T_H3K9me3_rep1_R1.fastq.gz H3K9me3_rep1
/path_to_file/1577752_ChIP_T_H3K4me3_rep2_R1.fastq.gz H3K4me3_rep2
/path_to_file/1577751_ChIP_T_H3K4me3_rep1_R1.fastq.gz H3K4me3_rep1
/path_to_file/1577750_ChIP_T_H3K4me1_rep2_R1.fastq.gz H3K4me1_rep2
/path_to_file/1577749_ChIP_T_H3K4me1_rep1_R1.fastq.gz H3K4me1_rep1
-pe: similar to fastq.tsv for paired-end data
3 columns: file_location for R1.fastq.gz, file_location for R2.fastq.gz, UID.
See an example below. This file is named as pe.list.
/path_to_file/1448387_T_R1_ATAC_R1.fastq.gz /path_to_file/1448387_T_R1_ATAC_R2.fastq.gz R1_ATAC
/path_to_file/1448388_T_R2_ATAC_R1.fastq.gz /path_to_file/1448388_T_R2_ATAC_R2.fastq.gz R2_ATAC
-d1: similar to peakcall.tsv for chip-seq data
For ChIP-seq data, usually you have an input control. For that, you want to use -d1.
Here, you want to compare everything to control, which could be input chip or IgG.
3 columns: UID, UID, label.
See an example below. This file is named as d1.tsv.
H3K36me3_rep2 input_rep2 H3K36me3
H3K36me3_rep1 input_rep1 H3K36me3
H3K27ac_rep2 input_rep2 H3K27ac
H3K27ac_rep1 input_rep1 H3K27ac
H3K27me3_rep2 input_rep2 H3K27me3
H3K27me3_rep1 input_rep1 H3K27me3
H3K9ac_rep2 input_rep2 H3K9ac
H3K9ac_rep1 input_rep1 H3K9ac
H3K9me3_rep2 input_rep2 H3K9me3
H3K9me3_rep1 input_rep1 H3K9me3
-d2: give your input files a label
For ATAC-seq data, you don’t have control. Then, use -d2.
Here, you want to state the label for your input files.
2 columns: UID, label.
See an example below. This file is named as d2.tsv.
R1_ATAC ATAC
R2_ATAC ATAC
Usage¶
Go to your data directory and type the following.
Step 0: Load python version 2.7.13.
module load python/2.7.13
Step 1: Prepare input parameters
chromHMM.py -pe PE_list -d1 design_matrix_1 -d2 design_matrix_2 -n 4
Tip
If files in -d1 are paired-end data, use --d1_bin_bam_addon " -paired". Similarly, if -d2 is paired-end data, use --d2_bin_bam_addon " -paired". See example below. This program can’t handle mix input in the same design matrix.
chromHMM.py -pe PE_list -d1 design_matrix_1 -d2 design_matrix_2 -n 4 --d1_bin_bam_addon " -paired" --d2_bin_bam_addon " -paired"
chromHMM.py -pe PE_list -d1 design_matrix_1 -d2 design_matrix_2 -n 25
For the specific example shown in the Input section, my command is:
[yli11@hpc01 chromHMM_pipeline]$ chromHMM.py -se se.list -pe pe.list -d1 d1.tsv -d2 d2.tsv -g hg38 --d2_bin_bam_addon " -paired" -m 200000
2019-07-15 17:35:12,714 - INFO - main - The job id is: chromHMM_yli11_2019-07-15
2019-07-15 17:35:12,714 - INFO - main - checking input files...
2019-07-15 17:35:12,715 - INFO - main - parsing se.list
2019-07-15 17:35:12,727 - INFO - main - parsing pe.list
2019-07-15 17:35:12,736 - INFO - main - All input files are found. Submitting jobs...
2019-07-15 17:35:12,742 - INFO - to_design_matrix - parsing d1.tsv
2019-07-15 17:35:12,758 - INFO - to_design_matrix - parsing d2.tsv
2019-07-15 17:35:12,843 - INFO - submit_pipeline_jobs - BWA_PE has been submitted; JobID: 83644249
2019-07-15 17:35:12,916 - INFO - submit_pipeline_jobs - BWA_SE has been submitted; JobID: 83644250
2019-07-15 17:35:13,160 - INFO - submit_pipeline_jobs - bin_bam has been submitted; JobID: 83644251
2019-07-15 17:35:13,666 - INFO - submit_pipeline_jobs - learn_model has been submitted; JobID: 83644252
2019-07-15 17:35:13,812 - INFO - submit_pipeline_jobs - infer_CS has been submitted; JobID: 83644253
2019-07-15 17:35:13,876 - INFO - submit_pipeline_jobs - email has been submitted; JobID: 83644254
Output¶
Once the job is finished, you will receive a notification email with the learned chromatin states attached (i.e., chromHMM_heatmap.pdf).
learned_states contains the chromHMM chromatin state discovery results, look at webpage_{{number_states}}.html for detailed description. _segments.bed contains the genome segments.
chromHMM_heatmap.pdf is the infered chromatin states, by comparing to the known associations (using euclidean distance). Note that row orders are sorted by chromatin state labels, which is not the same as the _segments.bed file.
Fine tuning chromHMM states¶
chromHMM itself is a dark art. Fine tuning is more like a personal taste. As long as the annotation is expected in some degree and you can link the results to some biological findings, then you don’t really need to worry too much about tuning the parameters. (Because more likely that, different parameters converged to the same or almost similar results.)
In this section, I have to mention one parameter to affect the binarizaiton significantly (chromHMM.jar binBam). Although I didn’t find it affect the actually learned states too much, I think tuning this parameter did increase the signal to noise ratio a little bit. This parameter is -p
poissonthreshold – This option specifies the tail probability of the poisson distribution that the binarization threshold should correspond to. The default value of this parameter is 0.0001.
I call it the binarization p-value. Basically, increase the p-value cutoff (default is 0.0001, change it to 1e-5) will decrease the number of 1s and vice versa.
In one example I have worked on, H3K4me1 occurs frequently in most of the learned states (looking at the emission probability), then we can increase the cutoff. Some TF chip-seq will have less occurrence then histone chip-seq, then we can lower the significance.
chromHMM.py -se default_histone_fastq.tsv -d1 default_histone_design.tsv -g mm9 -d_tf special_TF_design.tsv -tf special_TF_fastq.tsv -j p0000005_n8 -n 8 --design_matrix_His special_histone_design.tsv --His_fastq_list special_histone_fastq.tsv --adjust_histone_pvalue 0.0000005 --LearnModel_addon -u /path/chromHMM_input --adjust_TF_pvalue 0.005.
In this above command:
-se and -d1 are the fastq files applied to the default p-value, 0.0001.
-tf , -d_tf and --adjust_TF_pvalue are the ones applied to user-defined p-value.
--His_fastq_list, --design_matrix_His and --adjust_histone_pvalue are the ones applied to user-defined p-value.
-u gives the path to annotation bed files. Note that these bed files have to be stored in a folder named with the genome assembly. For example, suppose my current working dir is /path/cwd. Then I need to create /path/cwd/mm9/, and inside mm9 I can put some sorted 3-column chip-seq bed files or other kind of bed files. Then I have to use -u /path/cwd not /path/cwd/mm9.
Post Processing¶
This section is provided to solve the following problems:
Automatic chromHMM annotation may not be accurate. For example, you may not have H3K27me3 (a poised promoter state marker) but still get poised promoter states in your chromHMM_heatmap.pdf. In this case, you want to manually define the chromatin states
Note
Look at the computer generated chromHMM_heatmap.pdf, think about the best chromatin states before starting this analysis.
hpcf_interactive
cd {{jid}}/learned_model_{{N}}
cp reordered_row_annotation.txt new_names.txt
## edit new_names.txt
module load conda3
source activate /home/yli11/.conda/envs/py2
plot_chromHMM_emission_enrichment_heatmap.py -e emissions_{{N}}.txt -a myCellLine_{{N}}_overlap.txt -c new_names.txt -o some_name.pdf
An example of new_names.txt is shown below:
label r g b chromHMM_col_order
Active Promoter 1 255 0 0 8
Active Promoter 2 255 195 77 6
Weak Promoter 112 48 160 7
Active Enhancer 255 255 0 5
Weak Enhancer 0 128 0 4
Insulator 189 183 107 3
Heterochromatin 128 128 128 1
Heterochromatin 128 128 128 2
Note that the row order and RGB colors are the same as the result heatmap.pdf.
Create ppr chromHMM tracks for visualization.
Here we need a segmentation bed file from chromHMM.jar and an annotation file defined by user.
The row order in annotation.txt should be the same as the ones in chromHMM_segments.txt. And chromHMM uses E1, E2 for the state names. You can check out reordered_row_annotation.txt for the original chromHMM state names.
Heterochromatin Het 128 128 128
Heterochromatin Het 128 128 128
Insulator Ins 189 183 107
Weak Enhancer WeakE 255 195 77
Active Enhancer ActiveE 255 255 0
Active Promoter 2 ActiveP2 255 0 0
Weak Promoter WeakP 112 48 160
Active Promoter 1 ActiveP1 255 0 0
hpcf_interactive
dos2unix row_annotation.txt
module load htslib python/2.7.13
chromHMM_ppr_track.py -bed myCellLine_8_segments.bed -ann row_annotation.txt -o ppr_tracks.bed
The output *.sorted.gz* are ready to be uploaded to ppr.
Custom enrichment¶
Input_25_segments.bed is the learned states from chromHMM, you can get it from this pipeline.
other_bed is a folder containing all the bed files that you want to check for segment enrichments.
myOut is the output name.
module load java
chrHMM=/home/yli11/Programs/jar_tools/ChromHMM-1.18/ChromHMM.jar
java -jar $chrHMM OverlapEnrichment Input_25_segments.bed other_bed myOut
Example, adding R-loop info¶
# /research_jude/rgs01_jude/groups/chenggrp/projects/blood_regulome/chenggrp/HemPortal/HemTools_uniform_processed_files/Hudep2/Day0/chromHMM
[yli11@noderome203 chromHMM]$ head -n 2 se.list
H3K27ac_H2_D0_R1.fastq.gz H3K27ac_R1
H3K27me3_H2_D0_R2.fastq.gz H3K27me3_R2
[yli11@noderome203 chromHMM]$ head -n 2 pe.list
1345651_Hudep2_ATAC_1_R1.fastq.gz 1345651_Hudep2_ATAC_1_R2.fastq.gz ATAC_1
1345652_Hudep2_ATAC_2_R1.fastq.gz 1345652_Hudep2_ATAC_2_R2.fastq.gz ATAC_2
[yli11@noderome203 chromHMM]$ head -n 2 d1.tsv
H3K27ac_R1 Input_R1 H3K27ac
H3K27me3_R2 Input_R1 H3K27me3
[yli11@noderome203 chromHMM]$ head -n 2 d2.tsv
ATAC_1 ATAC
ATAC_2 ATAC
[yli11@noderome203 chromHMM]$ chromHMM.py -se se.list -pe pe.list -d1 d1.tsv -d2 d2.tsv -n 25 -g hg19 -m 250000 -j with_DRIP_25
2023-03-31 15:08:18,934 - INFO - main - The job id is: with_DRIP_25
2023-03-31 15:08:18,934 - INFO - main - checking input files...
2023-03-31 15:08:18,934 - INFO - main - parsing se.list
2023-03-31 15:08:18,954 - INFO - main - parsing pe.list
2023-03-31 15:08:18,970 - INFO - main - All input files are found. Submitting jobs...
2023-03-31 15:08:18,988 - INFO - to_design_matrix - parsing d1.tsv
2023-03-31 15:08:19,014 - INFO - to_design_matrix - parsing d2.tsv
2023-03-31 15:08:19,773 - INFO - submit_pipeline_jobs - BWA_PE has been submitted; JobID: 186766487
2023-03-31 15:08:19,845 - INFO - submit_pipeline_jobs - BWA_TF_SE has been submitted; JobID: 186766488
2023-03-31 15:08:19,930 - INFO - submit_pipeline_jobs - BWA_SE has been submitted; JobID: 186766489
2023-03-31 15:08:20,148 - INFO - submit_pipeline_jobs - BWA_His_SE has been submitted; JobID: 186766490
2023-03-31 15:08:20,218 - INFO - submit_pipeline_jobs - bin_bam has been submitted; JobID: 186766491
2023-03-31 15:08:20,688 - INFO - submit_pipeline_jobs - learn_model has been submitted; JobID: 186766492
2023-03-31 15:08:20,759 - INFO - submit_pipeline_jobs - infer_CS has been submitted; JobID: 186766493
2023-03-31 15:08:20,831 - INFO - submit_pipeline_jobs - email has been submitted; JobID: 186766494
Comments¶
code @ github.