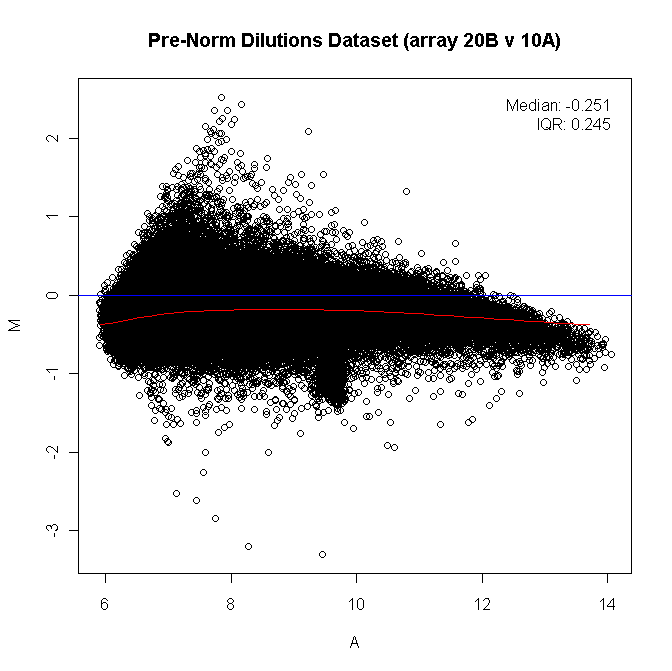
What is MA plot?¶
MA plot shows the log average (A) on the 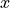 -axis and the log ratio (
-axis and the log ratio (M) on the 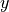 -axis. Here,
-axis. Here, M stands for minus because 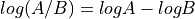
Similar plots are Bland-Altman plot, Tukey mean-difference plot, mean-difference plot, or MD plot.
This type of plot is good to show the data distribution between two individuals or two groups. Examples include:
show data distribution in two replicates or two groups to identify systematic bias (if normalization is needed)
show gene expression distribution comparing A to B, potentially highlighting differentially expressed genes, and/or other gene categories such as housekeeping genes, highly variable genes.
[50]:
import pandas as pd
import matplotlib.pylab as plt
import seaborn as sns
[51]:
df = pd.read_csv("/home/yli11/tmp/results.KO_vs_WT.csv",sep="\t",index_col=0)
df.head()
[51]:
| logFC | AveExpr | t | P.Value | adj.P.Val | B | WT_1_log2CPM | WT_2_log2CPM | WT_3_log2CPM | KO_1_log2CPM | KO_2_log2CPM | KO_3_log2CPM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene | ||||||||||||
| D17H6S56E-5 | -3.0830 | 9.3418 | -97.669 | 2.276800e-15 | 3.597600e-11 | 25.102 | 10.8880 | 10.9120 | 10.8500 | 7.7671 | 7.8119 | 7.8218 |
| Scd1 | -2.2133 | 6.1060 | -50.068 | 1.151200e-12 | 9.095200e-09 | 19.799 | 7.2574 | 7.1911 | 7.1920 | 5.0828 | 4.9264 | 4.9864 |
| Coro2a | -1.4558 | 7.9154 | -46.998 | 2.073900e-12 | 1.092300e-08 | 19.285 | 8.6433 | 8.6614 | 8.6256 | 7.1924 | 7.2202 | 7.1495 |
| Plxnb2 | -2.9373 | 3.6346 | -42.033 | 5.854300e-12 | 1.598600e-08 | 17.639 | 5.0743 | 5.1443 | 5.1107 | 2.2122 | 2.2622 | 2.0040 |
| Gzmb | -1.8469 | 4.9198 | -41.606 | 6.436800e-12 | 1.598600e-08 | 18.097 | 5.7934 | 5.8635 | 5.8686 | 3.9655 | 3.9610 | 4.0665 |
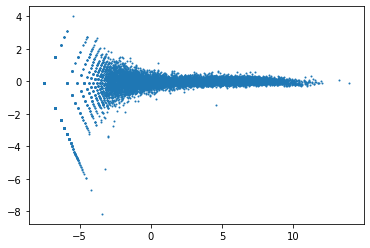
compare between replicates¶
[26]:
A = (df['WT_1_log2CPM']+df['WT_2_log2CPM'])/2
M = df['WT_1_log2CPM']-df['WT_2_log2CPM']
plt.scatter(x=A,y=M,s=1) # s is point size
[26]:
<matplotlib.collections.PathCollection at 0x2aad8f7f0280>
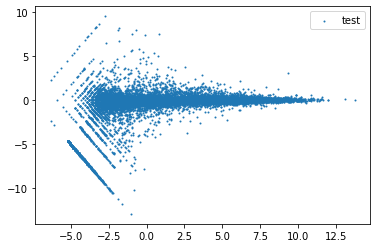
compare between two samples from different group¶
[59]:
A = (df['WT_1_log2CPM']+df['KO_1_log2CPM'])/2
M = df['WT_1_log2CPM']-df['KO_1_log2CPM']
plt.scatter(x=A,y=M,s=1) # s is point size
plt.legend(["test"])
[59]:
<matplotlib.legend.Legend at 0x2aad901dd6a0>
compare between groups¶
[20]:
# extract column names, you can also just type column names manually
WT_column_names = [x for x in df.columns if "WT" in x] # When using only 'if', put 'for' in the beginning
KO_column_names = [x for x in df.columns if "KO" in x] # When using only 'if', put 'for' in the beginning
print (WT_column_names)
print (KO_column_names)
['WT_1_log2CPM', 'WT_2_log2CPM', 'WT_3_log2CPM']
['KO_1_log2CPM', 'KO_2_log2CPM', 'KO_3_log2CPM']
[42]:
a=[1,2,3,"asd"]
a
[42]:
[1, 2, 3, 'asd']
[43]:
list(range(0,10))
[43]:
[0, 1, 2, 3, 4, 5, 6, 7, 8, 9]
[45]:
[i/2 for i in range(0,10)]
[45]:
[0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5]
[46]:
output = []
for i in range(0,10):
output.append(i/2)
output
[46]:
[0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5]
[47]:
df.columns
[47]:
Index(['logFC', 'AveExpr', 't', 'P.Value', 'adj.P.Val', 'B', 'WT_1_log2CPM',
'WT_2_log2CPM', 'WT_3_log2CPM', 'KO_1_log2CPM', 'KO_2_log2CPM',
'KO_3_log2CPM'],
dtype='object')
[48]:
df.shape
[48]:
(15801, 12)
[49]:
[i for i in df.columns if "W" in i]
[49]:
['WT_1_log2CPM', 'WT_2_log2CPM', 'WT_3_log2CPM']
[25]:
A = (df[WT_column_names].mean(axis=1)+df[KO_column_names].mean(axis=1))/2
M = df[KO_column_names].mean(axis=1)-df[WT_column_names].mean(axis=1)
plt.scatter(x=A,y=M,s=1) # s is point size
## add cosmetics
plt.title("KO vs WT")
plt.xlabel("log mean expression")
plt.ylabel("log fold change")
[25]:
Text(0, 0.5, 'log fold change')

[52]:
df.head()
[52]:
| logFC | AveExpr | t | P.Value | adj.P.Val | B | WT_1_log2CPM | WT_2_log2CPM | WT_3_log2CPM | KO_1_log2CPM | KO_2_log2CPM | KO_3_log2CPM | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gene | ||||||||||||
| D17H6S56E-5 | -3.0830 | 9.3418 | -97.669 | 2.276800e-15 | 3.597600e-11 | 25.102 | 10.8880 | 10.9120 | 10.8500 | 7.7671 | 7.8119 | 7.8218 |
| Scd1 | -2.2133 | 6.1060 | -50.068 | 1.151200e-12 | 9.095200e-09 | 19.799 | 7.2574 | 7.1911 | 7.1920 | 5.0828 | 4.9264 | 4.9864 |
| Coro2a | -1.4558 | 7.9154 | -46.998 | 2.073900e-12 | 1.092300e-08 | 19.285 | 8.6433 | 8.6614 | 8.6256 | 7.1924 | 7.2202 | 7.1495 |
| Plxnb2 | -2.9373 | 3.6346 | -42.033 | 5.854300e-12 | 1.598600e-08 | 17.639 | 5.0743 | 5.1443 | 5.1107 | 2.2122 | 2.2622 | 2.0040 |
| Gzmb | -1.8469 | 4.9198 | -41.606 | 6.436800e-12 | 1.598600e-08 | 18.097 | 5.7934 | 5.8635 | 5.8686 | 3.9655 | 3.9610 | 4.0665 |
[53]:
hk_genes = HemData.get_housekeeping_genes()
hk_genes
/home/yli11/.conda/envs/captureC/lib/python3.8/site-packages/HemTools/HemData.py:11: ParserWarning: Falling back to the 'python' engine because the 'c' engine does not support regex separators (separators > 1 char and different from '\s+' are interpreted as regex); you can avoid this warning by specifying engine='python'.
names = pd.read_csv(f"{pdir}/../HemData/index.tsv", sep="\s", header=None, index_col=0)[1].to_dict()
[53]:
| Mouse | Human | |
|---|---|---|
| 0 | 1600012H06Rik | C6orf120 |
| 1 | 1700123O20Rik | C14orf119 |
| 2 | 1810009A15Rik | C11orf98 |
| 3 | 1810013L24Rik | C16orf72 |
| 4 | 2610507B11Rik | KIAA0100 |
| ... | ... | ... |
| 1125 | Zmynd19 | ZMYND19 |
| 1126 | Zranb1 | ZRANB1 |
| 1127 | Zranb2 | ZRANB2 |
| 1128 | Zrsr1 | ZRSR2 |
| 1129 | Zyg11b | ZYG11B |
1130 rows × 2 columns
[ ]:
[56]:
A = (df[WT_column_names].mean(axis=1)+df[KO_column_names].mean(axis=1))/2
M = df[KO_column_names].mean(axis=1)-df[WT_column_names].mean(axis=1)
plt.scatter(x=A,y=M,s=1) # s is point size
# deg
deg = df[(df['logFC'].abs()>=2)&(df['adj.P.Val']<=0.01)]
degA = (deg[WT_column_names].mean(axis=1)+deg[KO_column_names].mean(axis=1))/2
degM = deg[KO_column_names].mean(axis=1)-deg[WT_column_names].mean(axis=1)
plt.scatter(x=degA,y=degM,s=3,color="red",label="DEG") # s is point size
from HemTools import HemData
hk_genes = HemData.get_housekeeping_genes()
# deg
hk = df.loc[df.index.intersection(hk_genes.Mouse)]
hkA = (hk[WT_column_names].mean(axis=1)+hk[KO_column_names].mean(axis=1))/2
hkM = hk[KO_column_names].mean(axis=1)-hk[WT_column_names].mean(axis=1)
plt.scatter(x=hkA,y=hkM,s=3,color="purple",label="housekeeping") # s is point size
## add cosmetics
plt.title("KO vs WT")
plt.xlabel("log mean expression")
plt.ylabel("log fold change")
plt.legend()
/home/yli11/.conda/envs/captureC/lib/python3.8/site-packages/HemTools/HemData.py:11: ParserWarning: Falling back to the 'python' engine because the 'c' engine does not support regex separators (separators > 1 char and different from '\s+' are interpreted as regex); you can avoid this warning by specifying engine='python'.
names = pd.read_csv(f"{pdir}/../HemData/index.tsv", sep="\s", header=None, index_col=0)[1].to_dict()
[56]:
<matplotlib.legend.Legend at 0x2aad9008ff10>
[ ]:
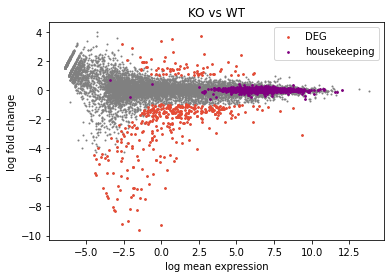
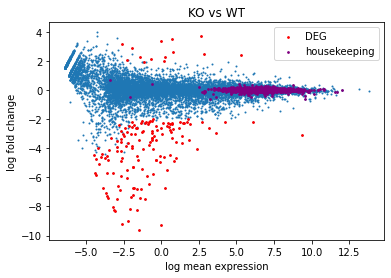
add more gene categories¶
differentially expressed genes
housekeeping genes
[41]:
A = (df[WT_column_names].mean(axis=1)+df[KO_column_names].mean(axis=1))/2
M = df[KO_column_names].mean(axis=1)-df[WT_column_names].mean(axis=1)
deg = df[(df['logFC'].abs()>=1)&(df['adj.P.Val']<=0.01)]
# Tip: use Ctrl+D (Windows) or Command + D (Mac) to do multi-selection and replace
degA = (deg[WT_column_names].mean(axis=1)+deg[KO_column_names].mean(axis=1))/2
degM = deg[KO_column_names].mean(axis=1)-deg[WT_column_names].mean(axis=1)
from HemTools import HemData
hk_genes = HemData.get_housekeeping_genes()
hk = df.loc[df.index.intersection(hk_genes.Mouse)]
# Tip: use Ctrl+D (Windows) or Command + D (Mac) to do multi-selection and replace
hkA = (hk[WT_column_names].mean(axis=1)+hk[KO_column_names].mean(axis=1))/2
hkM = hk[KO_column_names].mean(axis=1)-hk[WT_column_names].mean(axis=1)
plt.scatter(x=A,y=M,s=1,color="grey") # s is point size
plt.scatter(x=degA,y=degM,s=3,color="#E64B35",label="DEG") # s is point size
plt.scatter(x=hkA,y=hkM,s=3,color="purple",label="housekeeping") # s is point size
## add cosmetics
plt.title("KO vs WT")
plt.xlabel("log mean expression")
plt.ylabel("log fold change")
plt.legend()
/home/yli11/.conda/envs/captureC/lib/python3.8/site-packages/HemTools/HemData.py:11: ParserWarning: Falling back to the 'python' engine because the 'c' engine does not support regex separators (separators > 1 char and different from '\s+' are interpreted as regex); you can avoid this warning by specifying engine='python'.
names = pd.read_csv(f"{pdir}/../HemData/index.tsv", sep="\s", header=None, index_col=0)[1].to_dict()
[41]:
<matplotlib.legend.Legend at 0x2aad8ffe1e50>