Common NGS data formats and tools¶
Objectives¶
You should be familiar with the following common examples that we use daily:
Sequence retrieval
Read alignment
SAM/BAM query: index, sort, stats, visualization, filter, subset
SAM format, SAM flags
BED: sort, intersect (remove), merge
Data formats¶
fasta
fastq
bam / bam.bai / sam
bed / bed4 / bed6 / bed6+4 == narrowPeak / bed12
wiggle / bedgraph / bigwiggle
vcf
gtf / gff / gff3
tsv / csv
.py / .R / .sh / .jar / .pl
Please refer to the UCSC format FAQ for “official” definition
Tools¶
fastqc / fastp
seqtk
BWA / bowtie2 / STAR
samtools
bedtools
deeptools
scripting languange: Python
FASTA format¶
An example shown below.
;LCBO - Prolactin precursor - Bovine
; a sample sequence in FASTA format
; MDSKGSSQKGSRLLLLLVVSNLLLCQGVVSTPVCPNGPGNCQVSLRDLFDRAVMVSHYIHDLSS
EMFNEFDKRYAQGKGFITMALNSCHTSSLPTPEDKEQAQQTHHEVLMSLILGLLRSWNDPLYHL
VTEVRGMKGAPDAILSRAIEIEEENKRLLEGMEMIFGQVIPGAKETEPYPVWSGLPSLQTKDED
ARYSAFYNLLHCLRRDSSKIDTYLKLLNCRIIYNNNC*
>MCHU - Calmodulin - Human, rabbit, bovine, rat, and chicken
MADQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGNGTID
FPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLGEKLTDEEVDEMIREA
DIDGDGQVNYEEFVQMMTAK*
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
Syntax¶
“;” is for comments
“>” is for the name of a DNA/RNA/protein sequence
The sequence name can only be written in one line, no matter how long it is
The next line following “>” should not be an empty line
; invalid FASTA format
>Sequence_1
ACTTGG
; invalid FASTA format
>Sequence_2
>split line for names new sequence in human
ACGTGG
Other notes¶
IUPAC code for DNA letters:¶
IUPAC nucleotide code |
Base |
|---|---|
A |
Adenine |
C |
Cytosine |
G |
Guanine |
T (or U) |
Thymine (or Uracil) |
R |
A or G |
Y |
C or T |
S |
G or C |
W |
A or T |
K |
G or T |
M |
A or C |
B |
C or G or T |
D |
A or G or T |
H |
A or C or T |
V |
A or C or G |
N |
any base |
. or - |
gap |
Filename extension¶
Extension |
Meaning |
Notes |
|---|---|---|
fasta, fa |
generic FASTA |
Any generic fasta file. See below for other common FASTA file extensions |
fna |
FASTA nucleic acid |
Used generically to specify nucleic acids. |
ffn |
FASTA nucleotide of gene regions |
Contains coding regions for a genome. |
faa |
FASTA amino acid |
Contains amino acid sequences. A multiple protein fasta file can have the more specific extension mpfa. |
frn |
FASTA non-coding RNA |
Contains non-coding RNA regions for a genome, in DNA alphabet e.g. tRNA, rRNA |
Reference¶
See this wiki.
FASTA examples¶
1. Download human genome sequence from UCSC¶
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
ls
gunzip hg38.fa.gz
head hg38.fa
# How many chromosomes do we have?
grep ">" hg38.fa | wc -l
2. Sequence Retrieval using online tools¶
This is a common task. For example: given several genes, we want to extract gene promoters, or gene exons.
The question is simple, however, for many people who have never done it before, what we really need is a list of genomic coordinates.
Solution 1: Using ProteinPaint (https://proteinpaint.stjude.org/)¶
Fastest way for a single given genomic coordinate: e.g. chr11:5248160-5248251 or chr11 5248160 5248251
To get the reverse completement, just use some online tool, such as: https://www.bioinformatics.org/sms/rev_comp.html
Solution 2: Using Ensembl BioMart (https://www.ensembl.org/biomart/martview/)¶
Google ensembl biomart, click on the first hit.
Convenient way for getting promoter/UTR/exon/intron sequences given a list of gene names/IDs.
Exercise: get the promoter sequences for these genes
Hbg1
Hbg2
hbb
hba1
3: Sequence Retrieval using bedtools¶
The ultimate solution.
[6]:
!bedtools getfasta
Tool: bedtools getfasta (aka fastaFromBed)
Version: v2.30.0
Summary: Extract DNA sequences from a fasta file based on feature coordinates.
Usage: bedtools getfasta [OPTIONS] -fi <fasta> -bed <bed/gff/vcf>
Options:
-fi Input FASTA file
-fo Output file (opt., default is STDOUT
-bed BED/GFF/VCF file of ranges to extract from -fi
-name Use the name field and coordinates for the FASTA header
-name+ (deprecated) Use the name field and coordinates for the FASTA header
-nameOnly Use the name field for the FASTA header
-split Given BED12 fmt., extract and concatenate the sequences
from the BED "blocks" (e.g., exons)
-tab Write output in TAB delimited format.
-bedOut Report extract sequences in a tab-delimited BED format instead of in FASTA format.
- Default is FASTA format.
-s Force strandedness. If the feature occupies the antisense,
strand, the sequence will be reverse complemented.
- By default, strand information is ignored.
-fullHeader Use full fasta header.
- By default, only the word before the first space or tab
is used.
-rna The FASTA is RNA not DNA. Reverse complementation handled accordingly.
BED format¶
We are going to use a bed6 or a standard bed format, which contains 6 columns: chr, start, end, name, value, strand.
[8]:
!head CTCF.hg38.random.bed
chr6 92372038 92372057 JASPA_MA0139.1_CTCF_chr6_92372039 9.80328 -
chr1 113890670 113890689 JASPA_MA0139.1_CTCF_chr1_113890671 8.7541 -
chr16 12677107 12677126 JASPA_MA0139.1_CTCF_chr16_12677108 13.9836 -
chr11 116687055 116687074 JASPA_MA0139.1_CTCF_chr11_116687056 9.04918 -
chr13 32306850 32306869 JASPA_MA0139.1_CTCF_chr13_32306851 8.11475 +
chr4 55038826 55038845 JASPA_MA0139.1_CTCF_chr4_55038827 8.18033 +
chr4 116198084 116198103 JASPA_MA0139.1_CTCF_chr4_116198085 12.0328 -
chr12 24244394 24244413 JASPA_MA0139.1_CTCF_chr12_24244395 8.44262 -
chr8 47186476 47186495 JASPA_MA0139.1_CTCF_chr8_47186477 8.85246 +
chr4 82671394 82671413 JASPA_MA0139.1_CTCF_chr4_82671395 9.55738 -
BED3: A BED file where each feature is described by chrom, start, and end.
For example: chr1 11873 14409
BED4: A BED file where each feature is described by chrom, start, end, and name.
For example: chr1 11873 14409 uc001aaa.3
BED5: A BED file where each feature is described by chrom, start, end, name, and score.
For example: chr1 11873 14409 uc001aaa.3 0
BED6: A BED file where each feature is described by chrom, start, end, name, score, and strand.
For example: chr1 11873 14409 uc001aaa.3 0 +
BED12: A BED file where each feature is described by all twelve columns listed above.
For example: chr1 11873 14409 uc001aaa.3 0 + 11873 11873 0 3 354,109,1189, 0,739,1347
[12]:
!bedtools getfasta -fi hg38.fa -bed CTCF.hg38.random.bed -fo CTCF.fa ; head CTCF.fa
>chr6:92372038-92372057
TAGCTCCCTCTTCTGAAGC
>chr1:113890670-113890689
TCTTGCCACCCTCTGACCA
>chr16:12677107-12677126
GACTGGCACCTGCTGGGCA
>chr11:116687055-116687074
CACCTCCACCTCTAGGTCT
>chr13:32306850-32306869
aatccaggaggtggagatt
[13]:
!bedtools getfasta -fi hg38.fa -bed CTCF.hg38.random.bed -fo CTCF.fa -s; head CTCF.fa
>chr6:92372038-92372057(-)
GCTTCAGAAGAGGGAGCTA
>chr1:113890670-113890689(-)
TGGTCAGAGGGTGGCAAGA
>chr16:12677107-12677126(-)
TGCCCAGCAGGTGCCAGTC
>chr11:116687055-116687074(-)
AGACCTAGAGGTGGAGGTG
>chr13:32306850-32306869(+)
aatccaggaggtggagatt
[15]:
!bedtools getfasta -fi hg38.fa -bed CTCF.hg38.random.bed -fo CTCF.fa -s -name; head CTCF.fa
>JASPA_MA0139.1_CTCF_chr6_92372039::chr6:92372038-92372057(-)
GCTTCAGAAGAGGGAGCTA
>JASPA_MA0139.1_CTCF_chr1_113890671::chr1:113890670-113890689(-)
TGGTCAGAGGGTGGCAAGA
>JASPA_MA0139.1_CTCF_chr16_12677108::chr16:12677107-12677126(-)
TGCCCAGCAGGTGCCAGTC
>JASPA_MA0139.1_CTCF_chr11_116687056::chr11:116687055-116687074(-)
AGACCTAGAGGTGGAGGTG
>JASPA_MA0139.1_CTCF_chr13_32306851::chr13:32306850-32306869(+)
aatccaggaggtggagatt
[17]:
!bedtools getfasta -fi hg38.fa -bed CTCF.hg38.random.bed -fo CTCF.fa -s -nameOnly; head CTCF.fa
>JASPA_MA0139.1_CTCF_chr6_92372039(-)
GCTTCAGAAGAGGGAGCTA
>JASPA_MA0139.1_CTCF_chr1_113890671(-)
TGGTCAGAGGGTGGCAAGA
>JASPA_MA0139.1_CTCF_chr16_12677108(-)
TGCCCAGCAGGTGCCAGTC
>JASPA_MA0139.1_CTCF_chr11_116687056(-)
AGACCTAGAGGTGGAGGTG
>JASPA_MA0139.1_CTCF_chr13_32306851(+)
aatccaggaggtggagatt
[19]:
!bedtools getfasta -fi hg38.fa -bed CTCF.hg38.random.bed -fo CTCF.fa -s -nameOnly -tab; head CTCF.fa
JASPA_MA0139.1_CTCF_chr6_92372039(-) GCTTCAGAAGAGGGAGCTA
JASPA_MA0139.1_CTCF_chr1_113890671(-) TGGTCAGAGGGTGGCAAGA
JASPA_MA0139.1_CTCF_chr16_12677108(-) TGCCCAGCAGGTGCCAGTC
JASPA_MA0139.1_CTCF_chr11_116687056(-) AGACCTAGAGGTGGAGGTG
JASPA_MA0139.1_CTCF_chr13_32306851(+) aatccaggaggtggagatt
JASPA_MA0139.1_CTCF_chr4_55038827(+) aagtcagcaggtgccacaa
JASPA_MA0139.1_CTCF_chr4_116198085(-) ctgccacaagaggatacaa
JASPA_MA0139.1_CTCF_chr12_24244395(-) CAGTCAGTAGGAGGGGCTG
JASPA_MA0139.1_CTCF_chr8_47186477(+) taatcaccagtggtcagta
JASPA_MA0139.1_CTCF_chr4_82671395(-) tagcctgtagatgtcaatt
4: Sequence Retrieval using Python¶
The ultimate solution for more customized analyses.
PyRanges¶
[56]:
import pyranges as pr
import pandas as pd
[57]:
ctcf = pr.read_bed("CTCF.hg38.random.bed",as_df=False).sort() # sort will increase downstream analysis speed
ctcf.head()
[57]:
| Chromosome | Start | End | Name | Score | Strand | |
|---|---|---|---|---|---|---|
| 0 | chr1 | 11804507 | 11804526 | JASPA_MA0139.1_CTCF_chr1_11804508 | 13.27870 | + |
| 1 | chr1 | 29824616 | 29824635 | JASPA_MA0139.1_CTCF_chr1_29824617 | 9.62295 | + |
| 2 | chr1 | 23368354 | 23368373 | JASPA_MA0139.1_CTCF_chr1_23368355 | 15.57380 | - |
| 3 | chr1 | 29744978 | 29744997 | JASPA_MA0139.1_CTCF_chr1_29744979 | 8.57377 | - |
| 4 | chr1 | 113890670 | 113890689 | JASPA_MA0139.1_CTCF_chr1_113890671 | 8.75410 | - |
| 5 | chr1 | 116134359 | 116134378 | JASPA_MA0139.1_CTCF_chr1_116134360 | 11.63930 | - |
| 6 | chr1 | 153684245 | 153684264 | JASPA_MA0139.1_CTCF_chr1_153684246 | 8.45902 | - |
| 7 | chr1 | 223429627 | 223429646 | JASPA_MA0139.1_CTCF_chr1_223429628 | 10.93440 | - |
[58]:
type(ctcf)
[58]:
pyranges.pyranges.PyRanges
[59]:
ctcf.seq = pr.get_fasta(ctcf.unstrand(), "hg38.fa")
[60]:
ctcf.head()
[60]:
| Chromosome | Start | End | Name | Score | Strand | seq | |
|---|---|---|---|---|---|---|---|
| 0 | chr1 | 11804507 | 11804526 | JASPA_MA0139.1_CTCF_chr1_11804508 | 13.27870 | + | AGGCCAACAGAGGGCATAG |
| 1 | chr1 | 29824616 | 29824635 | JASPA_MA0139.1_CTCF_chr1_29824617 | 9.62295 | + | AGATCAGCAGAGGGCCCCT |
| 2 | chr1 | 23368354 | 23368373 | JASPA_MA0139.1_CTCF_chr1_23368355 | 15.57380 | - | CTCTGCCATCTCGAGGCCG |
| 3 | chr1 | 29744978 | 29744997 | JASPA_MA0139.1_CTCF_chr1_29744979 | 8.57377 | - | ccctgacctctgtaggtcc |
| 4 | chr1 | 113890670 | 113890689 | JASPA_MA0139.1_CTCF_chr1_113890671 | 8.75410 | - | TCTTGCCACCCTCTGACCA |
| 5 | chr1 | 116134359 | 116134378 | JASPA_MA0139.1_CTCF_chr1_116134360 | 11.63930 | - | GGATGCCACCTAATGGTGG |
| 6 | chr1 | 153684245 | 153684264 | JASPA_MA0139.1_CTCF_chr1_153684246 | 8.45902 | - | TCCTGCCGTCCAGTGGAGG |
| 7 | chr1 | 223429627 | 223429646 | JASPA_MA0139.1_CTCF_chr1_223429628 | 10.93440 | - | GCTTGCCCTCTGCAGCCCA |
Pyrange only works for unstranded dataframe
[61]:
def revcomp(seq):
tab = str.maketrans("ACTG", "TGAC")
return seq.translate(tab)[::-1]
ctcf.seq = ctcf.as_df().apply(lambda r: revcomp(r['seq']) if r['Strand']=="-" else r['seq'],axis=1)
[66]:
ctcf.head()
[66]:
| Chromosome | Start | End | Name | Score | Strand | seq | |
|---|---|---|---|---|---|---|---|
| 0 | chr1 | 11804507 | 11804526 | JASPA_MA0139.1_CTCF_chr1_11804508 | 13.27870 | + | AGGCCAACAGAGGGCATAG |
| 1 | chr1 | 29824616 | 29824635 | JASPA_MA0139.1_CTCF_chr1_29824617 | 9.62295 | + | AGATCAGCAGAGGGCCCCT |
| 2 | chr1 | 23368354 | 23368373 | JASPA_MA0139.1_CTCF_chr1_23368355 | 15.57380 | - | CGGCCTCGAGATGGCAGAG |
| 3 | chr1 | 29744978 | 29744997 | JASPA_MA0139.1_CTCF_chr1_29744979 | 8.57377 | - | cctggatgtctccagtccc |
| 4 | chr1 | 113890670 | 113890689 | JASPA_MA0139.1_CTCF_chr1_113890671 | 8.75410 | - | TGGTCAGAGGGTGGCAAGA |
| 5 | chr1 | 116134359 | 116134378 | JASPA_MA0139.1_CTCF_chr1_116134360 | 11.63930 | - | CCACCATTAGGTGGCATCC |
| 6 | chr1 | 153684245 | 153684264 | JASPA_MA0139.1_CTCF_chr1_153684246 | 8.45902 | - | CCTCCACTGGACGGCAGGA |
| 7 | chr1 | 223429627 | 223429646 | JASPA_MA0139.1_CTCF_chr1_223429628 | 10.93440 | - | TGGGCTGCAGAGGGCAAGC |
Look at sequence logo¶
[77]:
from Bio import motifs
from Bio.Seq import Seq
[82]:
m = motifs.create([Seq(x.upper()) for x in ctcf.seq.tolist()])
[84]:
m.weblogo("CTCF.png")
[86]:
from IPython.display import Image
Image("CTCF.png")
[86]:
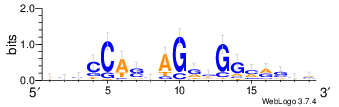
Manhattan plot¶
[108]:
from qmplot import manhattanplot
# https://github.com/ShujiaHuang/qmplot
manhattanplot(data=ctcf.as_df(),
chrom="Chromosome",
pos="Start",
pv="Score",
snp="Name",
logp=False,
ylabel="Score",
xlabel="Chromosome",
xticklabel_kws={"rotation": "vertical"},
is_show=True, # display the plot in screen
figname="output_manhattan_plot.png")
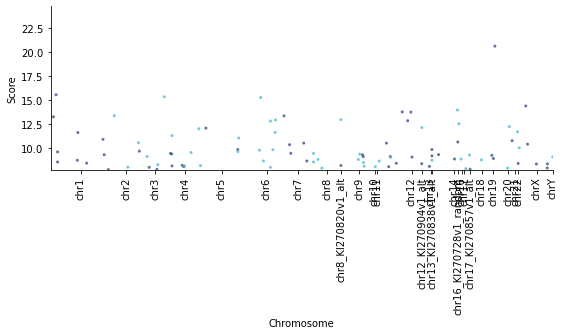
[108]:
<AxesSubplot:xlabel='Chromosome', ylabel='Score'>
FASTQ format¶
[110]:
!zcat test.10K.R1.fastq.gz | head
@M04990:66:000000000-G83PY:1:2101:2222:14575 1:N:0:TCTTCCTTTCCTTTTTTTGTTTTTCCTTCTTTCTCTTTCTTTTTTTTTGTTTTTTTTATTTGGTTTTTTTTTTTTTTTTCCTCTTGCATCTGTTGTTCT+TCTTTCCC
GCTCGACGCCATTAATAATGTTTTCCGTAAATTCAGCGCCTTCCATGATG
+
CCCDDDDCCCCCGGGGGGGGGGHHHHGHHGHHHHHHHGGGGGHHHHHHHH
@M04990:66:000000000-G83PY:1:1102:10573:9495 1:N:0:TCGTCCTCTCCTTGTGCCTGTTTCAATGCGTCCTCGCTCGCACTCACTTTTATCGAAGTGACTCGTTCGCCGTTGTGACACCTCGTCCACGCCAATTAT+TATCCTCT
CAATAGTCGTACGCCGATGCGAAACATCGGCCACGTGTGTCGATCTCGTA
+
CCCCBFFFCBFCGGGGFGGGGGGGGHHHGGGGGGGHHHHHHGHGFHGHGG
@M04990:66:000000000-G83PY:1:1102:15470:5522 1:N:0:CCCCGCCACCGTGGGGCGGGGTGGAAGCATGCGCCGCCCCCCGCCGGGCGGGGCGATGTTTTGTGGTAGCCGCGGCGCAAACACCCCCCCCACTAACCC+AGAGTAGA
CATTGGCACAGCTTGTCTCCAGGACCTTTTATTTTAGAACAAAAAAAAAA
gzip: stdout: Broken pipe
Syntax:¶
Each sequence/read is represented in 4 lines
first line is read name, followed by some descriptions
second line is the sequence
third line is just +
fourth line is the quality scores, same length as the sequence
Base qualities, increasing order:¶
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
FASTQ examples¶
1. take a look at the fastq files¶
Usually I just use less.
2. check sequencing quality¶
Use fastqc
[112]:
!mkdir test_fastqc
!module load fastqc; fastqc test.10K.R1.fastq.gz -o test_fastqc
Started analysis of test.10K.R1.fastq.gz
Approx 10% complete for test.10K.R1.fastq.gz
Approx 20% complete for test.10K.R1.fastq.gz
Approx 30% complete for test.10K.R1.fastq.gz
Approx 40% complete for test.10K.R1.fastq.gz
Approx 50% complete for test.10K.R1.fastq.gz
Approx 60% complete for test.10K.R1.fastq.gz
Approx 70% complete for test.10K.R1.fastq.gz
Approx 80% complete for test.10K.R1.fastq.gz
Approx 90% complete for test.10K.R1.fastq.gz
Approx 100% complete for test.10K.R1.fastq.gz
Analysis complete for test.10K.R1.fastq.gz
3. Random sampling fastq¶
[2]:
# sample N reads
!module load seqtk; seqtk sample test.10K.R1.fastq.gz 100 > test.sample.fastq;wc -l test.sample.fastq
400 test.sample.fastq
Note that seqtk only output uncompressed fastq, if you want to a fastq.gz file, you have to gzip it¶
[3]:
!module load seqtk; seqtk sample test.10K.R1.fastq.gz 100 > test.sample.fastq;gzip test.sample.fastq;zcat test.sample.fastq.gz|head
@M04990:66:000000000-G83PY:1:2104:15259:7127 1:N:0:TCGGACGATCATGGGTAGACGGACAAGTATGCAGCGCGCTCAAGCACGTGGATTACTTCGGAGTCGTACGCCGATGCGAAACATCGGCCACCAGATCTG+TAGATCGC
ATCCACAACTATCTGAAAAGGTTTATATTAAAATACTGCTCCCAGCCAGG
+
CCCCCFFCCFFFGGGGGGGGGGHHHHHHHHHHHHHHHHHHHHGHHHGHHG
@M04990:66:000000000-G83PY:1:2104:7163:24261 1:N:0:TCGGACGATCATGGGTAGACGGACAAGTATGCAGCGCGCTCAAGCACGTGGATTCTACGACAGTCGTACGCCGATGCGAAACATCGGCCACTTCCATTG+AGAGTAGA
GCCCATGCCCCAGCTCACCTCCATCATCGTCAACCAGCTCAAGAAGATCA
+
AACCAFFFFFCCGGGGGGGGGGHHHHHHHHHHGHGGHGHHHHHHHHHHHH
@M04990:66:000000000-G83PY:1:1103:27097:20242 1:N:0:TCGGACGATCATGGGTCTCACGGCAAGTATGCACGCGCTCAAGCACGTGGATTCTCACGGAGTCGTACGCCGATGCGAAACATCGGCCACGCATGGCTA+TAGATCGC
CTCCCAGGTTCACCTCATTTTCTTTGGCTCTTCAGTCCGCTGGTTTGAGT
[4]:
# Sample 10% reads
!module load seqtk; seqtk sample test.10K.R1.fastq.gz 0.1 > test.sample.fastq;wc -l test.sample.fastq
4000 test.sample.fastq
Also see: https://hemtools.readthedocs.io/en/latest/content/Bioinformatics_tools/fastqc.html for batch run
4. read alignment using BWA¶
[119]:
!module load bwa;bwa index hg38.fa
[bwa_index] Pack FASTA... 20.66 sec
[bwa_index] Construct BWT for the packed sequence...
[BWTIncCreate] textLength=6418572210, availableWord=463634060
[BWTIncConstructFromPacked] 10 iterations done. 99999986 characters processed.
[BWTIncConstructFromPacked] 20 iterations done. 199999986 characters processed.
[BWTIncConstructFromPacked] 30 iterations done. 299999986 characters processed.
[BWTIncConstructFromPacked] 40 iterations done. 399999986 characters processed.
[BWTIncConstructFromPacked] 50 iterations done. 499999986 characters processed.
[BWTIncConstructFromPacked] 60 iterations done. 599999986 characters processed.
[BWTIncConstructFromPacked] 70 iterations done. 699999986 characters processed.
[BWTIncConstructFromPacked] 80 iterations done. 799999986 characters processed.
[BWTIncConstructFromPacked] 90 iterations done. 899999986 characters processed.
[BWTIncConstructFromPacked] 100 iterations done. 999999986 characters processed.
[BWTIncConstructFromPacked] 110 iterations done. 1099999986 characters processed.
[BWTIncConstructFromPacked] 120 iterations done. 1199999986 characters processed.
[BWTIncConstructFromPacked] 130 iterations done. 1299999986 characters processed.
[BWTIncConstructFromPacked] 140 iterations done. 1399999986 characters processed.
[BWTIncConstructFromPacked] 150 iterations done. 1499999986 characters processed.
[BWTIncConstructFromPacked] 160 iterations done. 1599999986 characters processed.
[BWTIncConstructFromPacked] 170 iterations done. 1699999986 characters processed.
[BWTIncConstructFromPacked] 180 iterations done. 1799999986 characters processed.
[BWTIncConstructFromPacked] 190 iterations done. 1899999986 characters processed.
[BWTIncConstructFromPacked] 200 iterations done. 1999999986 characters processed.
[BWTIncConstructFromPacked] 210 iterations done. 2099999986 characters processed.
[BWTIncConstructFromPacked] 220 iterations done. 2199999986 characters processed.
[BWTIncConstructFromPacked] 230 iterations done. 2299999986 characters processed.
[BWTIncConstructFromPacked] 240 iterations done. 2399999986 characters processed.
[BWTIncConstructFromPacked] 250 iterations done. 2499999986 characters processed.
[BWTIncConstructFromPacked] 260 iterations done. 2599999986 characters processed.
[BWTIncConstructFromPacked] 270 iterations done. 2699999986 characters processed.
[BWTIncConstructFromPacked] 280 iterations done. 2799999986 characters processed.
[BWTIncConstructFromPacked] 290 iterations done. 2899999986 characters processed.
[BWTIncConstructFromPacked] 300 iterations done. 2999999986 characters processed.
[BWTIncConstructFromPacked] 310 iterations done. 3099999986 characters processed.
[BWTIncConstructFromPacked] 320 iterations done. 3199999986 characters processed.
[BWTIncConstructFromPacked] 330 iterations done. 3299999986 characters processed.
^C
[116]:
!module load bwa;\
bwa_index=/home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa;\
bwa mem $bwa_index test.10K.R1.fastq.gz test.10K.R2.fastq.gz > test.sam
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 20000 sequences (1000000 bp)...
[M::mem_pestat] # candidate unique pairs for (FF, FR, RF, RR): (0, 2289, 1, 0)
[M::mem_pestat] skip orientation FF as there are not enough pairs
[M::mem_pestat] analyzing insert size distribution for orientation FR...
[M::mem_pestat] (25, 50, 75) percentile: (62, 168, 228)
[M::mem_pestat] low and high boundaries for computing mean and std.dev: (1, 560)
[M::mem_pestat] mean and std.dev: (162.87, 115.26)
[M::mem_pestat] low and high boundaries for proper pairs: (1, 726)
[M::mem_pestat] skip orientation RF as there are not enough pairs
[M::mem_pestat] skip orientation RR as there are not enough pairs
[M::mem_process_seqs] Processed 20000 reads in 2.793 CPU sec, 2.793 real sec
[main] Version: 0.7.12-r1039
[main] CMD: bwa mem /home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa test.10K.R1.fastq.gz test.10K.R2.fastq.gz
[main] Real time: 22.389 sec; CPU: 4.152 sec
[187]:
!module avail bwa
------------------------ /hpcf/apps/modules/modulefiles ------------------------
bwa/0.5.10 bwa/0.7.5a bwa/0.7.15
bwa/0.6.1 bwa/0.7.12 (D) bwa/0.7.16a
---------------- /hpcf/authorized_apps/rhel7_apps/module_files -----------------
bwa/0.7.17
Where:
D: Default Module
Module defaults are chosen based on Find First Rules due to Name/Version/Version
modules found in the module tree.
See https://lmod.readthedocs.io/en/latest/060_locating.html for details.
Use "module spider" to find all possible modules and extensions.
Use "module keyword key1 key2 ..." to search for all possible modules matching
any of the "keys".
IMPORTANT
Notice that the bwa version is 0.7.12-r1039, but my index was created by 0.7.16a, so the aligned results may have some error. Better to use the same bwa version.
SAM Format¶
[122]:
!head -n 30 test.sam
@SQ SN:chr10 LN:133797422
@SQ SN:chr11 LN:135086622
@SQ SN:chr12 LN:133275309
@SQ SN:chr13 LN:114364328
@SQ SN:chr14 LN:107043718
@SQ SN:chr15 LN:101991189
@SQ SN:chr16 LN:90338345
@SQ SN:chr17 LN:83257441
@SQ SN:chr18 LN:80373285
@SQ SN:chr19 LN:58617616
@SQ SN:chr1 LN:248956422
@SQ SN:chr20 LN:64444167
@SQ SN:chr21 LN:46709983
@SQ SN:chr22 LN:50818468
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@SQ SN:chr4 LN:190214555
@SQ SN:chr5 LN:181538259
@SQ SN:chr6 LN:170805979
@SQ SN:chr7 LN:159345973
@SQ SN:chr8 LN:145138636
@SQ SN:chr9 LN:138394717
@SQ SN:chrM LN:16569
@SQ SN:chrX LN:156040895
@SQ SN:chrY LN:57227415
@PG ID:bwa PN:bwa VN:0.7.12-r1039 CL:bwa mem /home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa test.10K.R1.fastq.gz test.10K.R2.fastq.gz
M04990:66:000000000-G83PY:1:2101:2222:14575 77 * 0 0 * * 0 0 GCTCGACGCCATTAATAATGTTTTCCGTAAATTCAGCGCCTTCCATGATG CCCDDDDCCCCCGGGGGGGGGGHHHHGHHGHHHHHHHGGGGGHHHHHHHH AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:2101:2222:14575 141 * 0 0 * * 0 0 ATCATGAGTCAAGTTACTGAACAATCCGTACGTTTCCAGACCGCTTTGGC AABCCFFFFFFFGGGGGGGGGGHHHHHGGHGGHHHHHGHHGHGGGGGHHH AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:1102:10573:9495 77 * 0 0 * * 0 0 CAATAGTCGTACGCCGATGCGAAACATCGGCCACGTGTGTCGATCTCGTA CCCCBFFFCBFCGGGGFGGGGGGGGHHHGGGGGGGHHHHHHGHGFHGHGG AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:1102:10573:9495 141 * 0 0 * * 0 0 AACTACACAATCAACGACGCCCTCGTCTTCCTTCTCTTCCTTCCTGTCTC 1>111@@BD1F1FB11AE0AE00BA0AF021A2BG1GGFGFD1B111B2D AS:i:0 XS:i:0
For detailed SAM format specification, see the official reference: https://samtools.github.io/hts-specs/SAMv1.pdf¶
The header section¶
[126]:
!module load samtools/1.7;\
samtools view -H test.sam
@SQ SN:chr10 LN:133797422
@SQ SN:chr11 LN:135086622
@SQ SN:chr12 LN:133275309
@SQ SN:chr13 LN:114364328
@SQ SN:chr14 LN:107043718
@SQ SN:chr15 LN:101991189
@SQ SN:chr16 LN:90338345
@SQ SN:chr17 LN:83257441
@SQ SN:chr18 LN:80373285
@SQ SN:chr19 LN:58617616
@SQ SN:chr1 LN:248956422
@SQ SN:chr20 LN:64444167
@SQ SN:chr21 LN:46709983
@SQ SN:chr22 LN:50818468
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@SQ SN:chr4 LN:190214555
@SQ SN:chr5 LN:181538259
@SQ SN:chr6 LN:170805979
@SQ SN:chr7 LN:159345973
@SQ SN:chr8 LN:145138636
@SQ SN:chr9 LN:138394717
@SQ SN:chrM LN:16569
@SQ SN:chrX LN:156040895
@SQ SN:chrY LN:57227415
@PG ID:bwa PN:bwa VN:0.7.12-r1039 CL:bwa mem /home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa test.10K.R1.fastq.gz test.10K.R2.fastq.gz
@SQ Reference sequence dictionary. The order of @SQ lines defines the alignment sorting order.
SN: Reference sequence name
LN: Reference sequence length
The alignment section¶
[127]:
!module load samtools/1.7;\
samtools view test.sam | head
M04990:66:000000000-G83PY:1:2101:2222:14575 77 * 0 0 * * 0 0 GCTCGACGCCATTAATAATGTTTTCCGTAAATTCAGCGCCTTCCATGATG CCCDDDDCCCCCGGGGGGGGGGHHHHGHHGHHHHHHHGGGGGHHHHHHHH AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:2101:2222:14575 141 * 0 0 * * 0 0 ATCATGAGTCAAGTTACTGAACAATCCGTACGTTTCCAGACCGCTTTGGC AABCCFFFFFFFGGGGGGGGGGHHHHHGGHGGHHHHHGHHGHGGGGGHHH AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:1102:10573:9495 77 * 0 0 * * 0 0 CAATAGTCGTACGCCGATGCGAAACATCGGCCACGTGTGTCGATCTCGTA CCCCBFFFCBFCGGGGFGGGGGGGGHHHGGGGGGGHHHHHHGHGFHGHGG AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:1102:10573:9495 141 * 0 0 * * 0 0 AACTACACAATCAACGACGCCCTCGTCTTCCTTCTCTTCCTTCCTGTCTC 1>111@@BD1F1FB11AE0AE00BA0AF021A2BG1GGFGFD1B111B2D AS:i:0 XS:i:0
M04990:66:000000000-G83PY:1:1102:15470:5522 83 chr7 113829330 0 21M29S = 113829329 -22 TTTTTTTTTTGTTCTAAAATAAAAGGTCCTGGAGACAAGCTGTGCCAATG GEAECGA0GFHFAFHGHHGGGHHHGGC11BGGFGGGGGAFFFFC1A@AAA NM:i:0 MD:Z:21 AS:i:21 XS:i:21 XA:Z:chr12,+60139811,26S21M3S,0;chr2,-139726748,3S20M27S,0;chr7,+88270110,23S19M8S,0;
M04990:66:000000000-G83PY:1:1102:15470:5522 163 chr7 113829329 0 16S22M12S = 113829330 22 ATGTAAGGGGTTTTTTTTTTTTTTTTTGTTCTAAAATAAAAGGTCCTGGA AABAAFFFFBBBGFGGGGGGGGGGGGG0BF3@BFFFEGHGB3EBFE3GF2 NM:i:0 MD:Z:22 AS:i:22 XS:i:25
M04990:66:000000000-G83PY:1:1103:16433:22048 65 chrX 24701555 0 50M chr9 92816867 0 CCTCCCAAGTAGCTGGGATTACAGGTGTCTGCCACCACGCCTGGCTGATT CCCCCCFFCFFFGGGGGGGGGGHHHHHHHHHHHHHHHHGGGHHHHHGHHH NM:i:1 MD:Z:46A3 AS:i:46 XS:i:46
M04990:66:000000000-G83PY:1:1103:16433:22048 129 chr9 92816867 0 50M chrX 24701555 0 CTGTAATCCCAGTACTTTGGGGGGCTGAGATGGGTGGATCATGAGGTCAG BBBBBFFFFFFFGCGGGGGGGGGGGGGHHGHHHHGEGHHHHHHGHFHGHH NM:i:2 MD:Z:12C8A28 AS:i:40 XS:i:40
M04990:66:000000000-G83PY:1:2104:26463:8539 99 chr8 113276127 60 50M = 113276316 239 GCATGAAATTGTAGGCCAAATAATGCCCCTCTACCTCCCCAAAAATGTAT BBBBBFFFFFFFGGC4GGGGGGHFFHHHGHGHHHHHHHGGHGHHHHHHHH NM:i:0 MD:Z:50 AS:i:50 XS:i:0
M04990:66:000000000-G83PY:1:2104:26463:8539 147 chr8 113276316 60 50M = 113276127 -239 AGAAATTTCTTCACCTGGAGTATAAGATGTGTGGAGGGAGAAAGTAGCAG HHHHHHGHHHHHHHHHHHHHHHHHHHHGGGGGGGGGGGFFFFFFFBBBBB NM:i:0 MD:Z:50 AS:i:50 XS:i:19
samtools view: writing to standard output failed: Broken pipe
samtools view: error closing standard output: -1
Col |
Field |
Type |
N/A Value |
Description |
|---|---|---|---|---|
1 |
QNAME |
string |
mandatory |
The query/read name. |
2 |
FLAG |
int |
mandatory |
The record’s flag. |
3 |
RNAME |
string |
* |
The reference name. |
4 |
POS |
32-bit int |
0 |
1-based position on the reference. |
5 |
MAPQ |
8-bit int |
255 |
The mapping quality. |
6 |
CIGAR |
string |
* |
The CIGAR string of the alignment. |
7 |
RNEXT |
string |
* |
The reference of the next mate/segment. |
8 |
PNEXT |
string |
0 |
The position of the next mate/seqgment. |
9 |
TLEN |
string |
0 |
The observed length of the template. |
10 |
SEQ |
string |
* |
The query/read sequence. |
11 |
QUAL |
string |
* |
The ASCII PHRED-encoded base qualities. |
Notes:
The SAM standard talks about “queries”. In the context of read mapping, where the format originates, queries are reads.
The SAM standard talks about “templates” and “segments”. In the case of paired-end and mate-pair mapping the template consists of two segments, each is one read. The template length is the insert size.
Paired-end reads are stored as two alignments records with the same QNAME. The first and second mate are discriminated by the FLAG values.
When the FLAG indicates that SEQ is reverse-complemented, then QUAL is reversed.
Positions in the SAM file are 1-based.
The qualities must be stored as ASCII PRED-encoded qualities.
The query and reference names must not contain whitespace. It is common to trim query and reference ids at the first space.
Getting the exact mapping coordinates directly based on the 4th column is not trivial, better to use bedtools for correctness.
MAPQ 0 could mean multi-mapped or repeat regions.
SAM flags¶
I usually use this online tool to make sense of SAM flags: https://broadinstitute.github.io/picard/explain-flags.html
BAM format¶
Binary format for SAM files. An index file (.bai) is usually generated for each position sorted BAM file
Common Examples¶
1. Convert SAM to BAM¶
2. Sort BAM file¶
3. Index BAM file¶
4. Get summary statistics¶
[131]:
!module load samtools/1.7;samtools view -b test.sam > test.bam
[132]:
!module load samtools/1.7;samtools sort -o test.st.bam test.bam
[133]:
!module load samtools/1.7;samtools index test.st.bam
[134]:
!module load samtools/1.7;samtools flagstat test.st.bam
20000 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
8455 + 0 mapped (42.27% : N/A)
20000 + 0 paired in sequencing
10000 + 0 read1
10000 + 0 read2
7854 + 0 properly paired (39.27% : N/A)
7994 + 0 with itself and mate mapped
461 + 0 singletons (2.31% : N/A)
62 + 0 with mate mapped to a different chr
23 + 0 with mate mapped to a different chr (mapQ>=5)
[135]:
!module load samtools/1.7;samtools idxstats test.st.bam
chr10 133797422 277 22
chr11 135086622 326 22
chr12 133275309 307 22
chr13 114364328 143 14
chr14 107043718 215 16
chr15 101991189 169 8
chr16 90338345 238 11
chr17 83257441 261 14
chr18 80373285 116 4
chr19 58617616 312 13
chr1 248956422 681 36
chr20 64444167 160 3
chr21 46709983 413 39
chr22 50818468 115 7
chr2 242193529 629 40
chr3 198295559 410 27
chr4 190214555 311 15
chr5 181538259 397 22
chr6 170805979 541 23
chr7 159345973 356 18
chr8 145138636 520 24
chr9 138394717 295 21
chrM 16569 1029 28
chrX 156040895 196 10
chrY 57227415 38 2
* 0 0 11084
[7]:
!module load samtools/1.7;samtools stats test.st.bam | head -n 50
# This file was produced by samtools stats (1.7+htslib-1.7) and can be plotted using plot-bamstats
# This file contains statistics for all reads.
# The command line was: stats test.st.bam
# CHK, Checksum [2]Read Names [3]Sequences [4]Qualities
# CHK, CRC32 of reads which passed filtering followed by addition (32bit overflow)
CHK fae7cc8c e08cae0b f53795c5
# Summary Numbers. Use `grep ^SN | cut -f 2-` to extract this part.
SN raw total sequences: 20000
SN filtered sequences: 0
SN sequences: 20000
SN is sorted: 1
SN 1st fragments: 10000
SN last fragments: 10000
SN reads mapped: 8455
SN reads mapped and paired: 7994 # paired-end technology bit set + both mates mapped
SN reads unmapped: 11545
SN reads properly paired: 7854 # proper-pair bit set
SN reads paired: 20000 # paired-end technology bit set
SN reads duplicated: 0 # PCR or optical duplicate bit set
SN reads MQ0: 1325 # mapped and MQ=0
SN reads QC failed: 0
SN non-primary alignments: 0
SN total length: 1000000 # ignores clipping
SN bases mapped: 422750 # ignores clipping
SN bases mapped (cigar): 385042 # more accurate
SN bases trimmed: 0
SN bases duplicated: 0
SN mismatches: 1936 # from NM fields
SN error rate: 5.028023e-03 # mismatches / bases mapped (cigar)
SN average length: 50
SN maximum length: 50
SN average quality: 33.5
SN insert size average: 183.9
SN insert size standard deviation: 140.8
SN inward oriented pairs: 3161
SN outward oriented pairs: 168
SN pairs with other orientation: 2
SN pairs on different chromosomes: 31
# First Fragment Qualitites. Use `grep ^FFQ | cut -f 2-` to extract this part.
# Columns correspond to qualities and rows to cycles. First column is the cycle number.
FFQ 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 49 0 36 0 0 0 0 0 0 0 0 0 0 164 27 6 1898 3254 3603 750 213 0 0 0 0 0
FFQ 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 48 0 27 0 0 0 0 0 0 0 0 0 0 118 40 33 1529 2971 3837 1061 336 0 0 0 0 0
FFQ 3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 41 0 12 0 0 0 0 0 0 0 0 0 0 83 41 28 1311 2898 3895 1263 428 0 0 0 0 0
FFQ 4 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 45 0 26 0 0 0 0 0 0 0 0 0 0 66 29 35 1229 2838 3858 1394 480 0 0 0 0 0
FFQ 5 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 79 0 22 0 0 0 0 0 0 0 0 0 0 72 55 39 1212 2759 3904 1392 466 0 0 0 0 0
FFQ 6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 52 0 15 6 8 0 0 0 0 0 0 0 0 26 20 43 210 784 1074 571 129 7062 0 0 0 0
FFQ 7 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 59 0 18 10 7 0 0 0 0 0 0 0 0 23 19 49 239 768 1023 597 132 7056 0 0 0 0
FFQ 8 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 56 5 18 9 12 0 0 0 0 0 0 0 0 24 22 52 238 829 1137 632 154 6812 0 0 0 0
FFQ 9 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 68 6 18 14 9 0 0 0 0 0 0 0 0 30 28 47 246 821 1160 711 182 6660 0 0 0 0
FFQ 10 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 60 7 16 19 10 0 0 0 0 0 0 0 0 38 21 63 205 833 1177 788 203 6558 0 0 0 0
For mapping multiple samples, use the bwa_mem pipeline¶
https://hemtools.readthedocs.io/en/latest/content/NGS_pipelines/bwa_mem.html
module load python/2.7.13
run_lsf.py --guess_input
run_lsf.py -f fastq.tsv -p bwa_mem -g mm9
5. visualize bam file in IGV¶
reads are sub-sampled
highligh a specific read
view as pairs
sort reads by mapping start
6. remove PCR duplicates (rmdup or dedup)¶
What is PCR duplicates? Simply put, identical reads. More precisely, the positions on 5’-end are identical. (that means 3’-end can be different)
For single-end sequencing, rmdup is usually not performed.
For paired-end sequencing, rmdup may be performed.
We never perform rmdup for RNA-seq or amplicon sequencing data.
[148]:
!module load samtools/1.7; \
samtools sort -n -o test.name.st.bam test.st.bam; \
samtools fixmate -m test.name.st.bam test.fixmate.bam; \
samtools sort -o test.fixmate.st.bam test.fixmate.bam; \
samtools markdup test.fixmate.st.bam test.markdup.st.bam; \
samtools markdup -r -s test.markdup.st.bam test.rmdup.st.bam
READ 20000 WRITTEN 19985
EXCLUDED 11545 EXAMINED 8455
PAIRED 7994 SINGLE 461
DULPICATE PAIR 10 DUPLICATE SINGLE 5
DUPLICATE TOTAL 15
[152]:
!module load samtools/1.7; samtools view -H test.fixmate.bam
@HD VN:1.5 SO:queryname
@SQ SN:chr10 LN:133797422
@SQ SN:chr11 LN:135086622
@SQ SN:chr12 LN:133275309
@SQ SN:chr13 LN:114364328
@SQ SN:chr14 LN:107043718
@SQ SN:chr15 LN:101991189
@SQ SN:chr16 LN:90338345
@SQ SN:chr17 LN:83257441
@SQ SN:chr18 LN:80373285
@SQ SN:chr19 LN:58617616
@SQ SN:chr1 LN:248956422
@SQ SN:chr20 LN:64444167
@SQ SN:chr21 LN:46709983
@SQ SN:chr22 LN:50818468
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@SQ SN:chr4 LN:190214555
@SQ SN:chr5 LN:181538259
@SQ SN:chr6 LN:170805979
@SQ SN:chr7 LN:159345973
@SQ SN:chr8 LN:145138636
@SQ SN:chr9 LN:138394717
@SQ SN:chrM LN:16569
@SQ SN:chrX LN:156040895
@SQ SN:chrY LN:57227415
@PG ID:bwa PN:bwa VN:0.7.12-r1039 CL:bwa mem /home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa test.10K.R1.fastq.gz test.10K.R2.fastq.gz
[150]:
!module load samtools/1.7; samtools view -H test.rmdup.st.bam
@HD VN:1.5 SO:coordinate
@SQ SN:chr10 LN:133797422
@SQ SN:chr11 LN:135086622
@SQ SN:chr12 LN:133275309
@SQ SN:chr13 LN:114364328
@SQ SN:chr14 LN:107043718
@SQ SN:chr15 LN:101991189
@SQ SN:chr16 LN:90338345
@SQ SN:chr17 LN:83257441
@SQ SN:chr18 LN:80373285
@SQ SN:chr19 LN:58617616
@SQ SN:chr1 LN:248956422
@SQ SN:chr20 LN:64444167
@SQ SN:chr21 LN:46709983
@SQ SN:chr22 LN:50818468
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@SQ SN:chr4 LN:190214555
@SQ SN:chr5 LN:181538259
@SQ SN:chr6 LN:170805979
@SQ SN:chr7 LN:159345973
@SQ SN:chr8 LN:145138636
@SQ SN:chr9 LN:138394717
@SQ SN:chrM LN:16569
@SQ SN:chrX LN:156040895
@SQ SN:chrY LN:57227415
@PG ID:bwa PN:bwa VN:0.7.12-r1039 CL:bwa mem /home/yli11/Data/Human/hg38/bwa_16a_index/hg38.fa test.10K.R1.fastq.gz test.10K.R2.fastq.gz
7. remove multi-mapped reads (uq)¶
[153]:
!module load samtools/1.7; samtools view -q 1 -b test.rmdup.st.bam > test.rmdup.uq.st.bam
8. filter reads by SAM flags¶
-f INT only include reads with all of the FLAGs in INT present [0]
-F INT only include reads with none of the FLAGS in INT present [0]
-f 2 -F 4 -F 8 -F 256 -F 512 -F 2048¶
[155]:
!module load samtools/1.7; samtools view -f 2 -F 4 -F 8 -F 256 -F 512 -F 2048 -b test.rmdup.uq.st.bam > test.valid_pairs.rmdup.uq.st.bam
9.1 subset SAM/BAM files using samtools view based on coordinates¶
[159]:
# subset by a single region
!module load samtools/1.7; samtools index test.valid_pairs.rmdup.uq.st.bam;\
samtools view test.valid_pairs.rmdup.uq.st.bam "chr10:610000-660000"
M04990:66:000000000-G83PY:1:2101:19247:11601 163 chr10 613723 60 50M = 613849 176 CCTCTCCACACCCTGGTAAACATGGCATTTCGCACACCTGCTTTGGTCCC >AABAFFFFFDDGGGGGGGGGGHHHHHHHHHHGGGGHGHHHHHHGHHHHH NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1872
M04990:66:000000000-G83PY:1:2101:19247:11601 83 chr10 613849 60 50M = 613723 -176 TTCCCCGGCCACCCTCCTGATCTCGGCCATCTGATGGCATCACTCCCCAC GGGGGGHHGGGGGGHHHHHGGGGGHHHHGGGGGGGGGGFFFBCCDDCCBC NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1879
[160]:
# subset by a bed file
!module load samtools/1.7; \
samtools view test.valid_pairs.rmdup.uq.st.bam -L ctcfpos.bed
M04990:66:000000000-G83PY:1:1101:17665:21940 83 chr6 30617302 60 50M = 30617169 -183 CGTGCTAGTGAAACGCCCTTGTCGCGAGACATTATACGCGAGGCGTGAAC GHHHHHHHHFGGGGGGHHGGGGGFGGHHGGGGGGGGGGBCFCFBFBCCCC NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1855
M04990:66:000000000-G83PY:1:2104:3420:8655 163 chr6 36842224 60 50M = 36842603 429 CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA >>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2 NM:i:0 MD:Z:50 AS:i:50 XS:i:19 MQ:i:40 MC:Z:50M ms:i:1875
9.2 subset SAM/BAM files using bedtools intersect based on coordinates¶
[162]:
!module load bedtools; \
bedtools intersect -a test.valid_pairs.rmdup.uq.st.bam -b ctcfpos.bed | samtools view
M04990:66:000000000-G83PY:1:1101:17665:21940 83 chr6 30617302 60 50M = 30617169 -183 CGTGCTAGTGAAACGCCCTTGTCGCGAGACATTATACGCGAGGCGTGAAC GHHHHHHHHFGGGGGGHHGGGGGFGGHHGGGGGGGGGGBCFCFBFBCCCC NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1855
M04990:66:000000000-G83PY:1:2104:3420:8655 163 chr6 36842224 60 50M = 36842603 429 CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA >>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2 NM:i:0 MD:Z:50 AS:i:50 XS:i:19 MQ:i:40 MC:Z:50M ms:i:1875
[169]:
!module load bedtools; \
bedtools intersect -a test.valid_pairs.rmdup.uq.st.bam -b ctcfpos.bed | bedtools bamtofastq -i - -fq test.fq | head test.fq
@M04990:66:000000000-G83PY:1:1101:17665:21940
GTTCACGCCTCGCGTATAATGTCTCGCGACAAGGGCGTTTCACTAGCACG
+
CCCCBFBFCFCBGGGGGGGGGGHHGGFGGGGGHHGGGGGGFHHHHHHHHG
@M04990:66:000000000-G83PY:1:2104:3420:8655
CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA
+
>>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2
9.3 subset SAM/BAM files using picard based on read list¶
[176]:
!echo M04990:66:000000000-G83PY:1:1101:17665:21940 > test.list
!echo M04990:66:000000000-G83PY:1:2104:3420:8655 >> test.list
!head test.list
M04990:66:000000000-G83PY:1:1101:17665:21940
M04990:66:000000000-G83PY:1:2104:3420:8655
[177]:
!module load java/10.0.2;\
java -jar /hpcf/apps/picard/install/2.9.4/picard.jar\
FilterSamReads I=test.valid_pairs.rmdup.uq.st.bam O=test.out.bam READ_LIST_FILE=test.list FILTER=includeReadList;\
module load samtools;samtools view test.out.bam
Picked up _JAVA_OPTIONS: -Djava.io.tmpdir=/research/rgs01/scratch_lsf/java -XX:ParallelGCThreads=1
[Tue Aug 17 00:31:03 CDT 2021] picard.sam.FilterSamReads INPUT=test.valid_pairs.rmdup.uq.st.bam FILTER=includeReadList [OUTPUT SAM/BAM will contain reads that are supplied in the READ_LIST_FILE file] READ_LIST_FILE=test.list OUTPUT=test.out.bam WRITE_READS_FILES=true VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false GA4GH_CLIENT_SECRETS=client_secrets.json
[Tue Aug 17 00:31:03 CDT 2021] Executing as yli11@noderome182 on Linux 3.10.0-1160.15.2.el7.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 10.0.2+13; Picard version: 2.9.4-SNAPSHOT
INFO 2021-08-17 00:31:03 FilterSamReads Filtering [presorted=true] test.valid_pairs.rmdup.uq.st.bam -> OUTPUT=test.out.bam [sortorder=coordinate]
INFO 2021-08-17 00:31:03 FilterSamReads 4 SAMRecords written to test.out.bam
[Tue Aug 17 00:31:03 CDT 2021] picard.sam.FilterSamReads done. Elapsed time: 0.00 minutes.
Runtime.totalMemory()=2147483648
M04990:66:000000000-G83PY:1:1101:17665:21940 163 chr6 30617169 60 50M = 30617302 183 ATCCAACGCTCTCCGCAAAATTTTACCCACTAGACGACAAAGTAGGCAAA 3>AABFFCCCCDGGGGGGGGGGHHHHHHHGHHHHHGGGGGHHHHHHHHHH NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1868
M04990:66:000000000-G83PY:1:1101:17665:21940 83 chr6 30617302 60 50M = 30617169 -183 CGTGCTAGTGAAACGCCCTTGTCGCGAGACATTATACGCGAGGCGTGAAC GHHHHHHHHFGGGGGGHHGGGGGFGGHHGGGGGGGGGGBCFCFBFBCCCC NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1855
M04990:66:000000000-G83PY:1:2104:3420:8655 163 chr6 36842224 60 50M = 36842603 429 CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA >>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2 NM:i:0 MD:Z:50 AS:i:50 XS:i:19 MQ:i:40 MC:Z:50M ms:i:1875
M04990:66:000000000-G83PY:1:2104:3420:8655 83 chr6 36842603 40 50M = 36842224 -429 TGGGAGGCAGAGGTTGCAGTGAGCCGAGATCGCACCACTGCACTCCAGCC HGHHHHHHHHHHHHHHBHHFGGGGHHHGGGFGFGGGGGFFFFFFBBBBBA NM:i:0 MD:Z:50 AS:i:50 XS:i:50 MQ:i:60 MC:Z:50M ms:i:1708
10. Bam to fastq¶
[178]:
!module load bedtools;bedtools bamtofastq -i test.valid_pairs.rmdup.uq.st.bam -fq test.fq | head test.fq
@M04990:66:000000000-G83PY:1:1101:17665:21940
GTTCACGCCTCGCGTATAATGTCTCGCGACAAGGGCGTTTCACTAGCACG
+
CCCCBFBFCFCBGGGGGGGGGGHHGGFGGGGGHHGGGGGGFHHHHHHHHG
@M04990:66:000000000-G83PY:1:2104:3420:8655
CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA
+
>>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2
get fastq using seqtk¶
[184]:
# Here, we are missing the index reads, because they are not part of the read names
# If we want to take a look at the index reads, we can use seqtk to subset fastq files
# Also, sometimes the input fastq to BWA mem is pre-processed (i.e., trimed), so if
# we want to see the raw reads, we need to subset raw fastq
!module load bedtools; \
bedtools intersect -a test.valid_pairs.rmdup.uq.st.bam -b ctcfpos.bed | samtools view
M04990:66:000000000-G83PY:1:1101:17665:21940 83 chr6 30617302 60 50M = 30617169 -183 CGTGCTAGTGAAACGCCCTTGTCGCGAGACATTATACGCGAGGCGTGAAC GHHHHHHHHFGGGGGGHHGGGGGFGGHHGGGGGGGGGGBCFCFBFBCCCC NM:i:0 MD:Z:50 AS:i:50 XS:i:0 MQ:i:60 MC:Z:50M ms:i:1855
M04990:66:000000000-G83PY:1:2104:3420:8655 163 chr6 36842224 60 50M = 36842603 429 CACATGCTGTGTGCTGGGTGCAGTCCTAGGCTCAGAGAATAAAAGGCAAA >>A3>BFFFFFFGGGGC2GEA4DFGHHHCBAGHHFFBFFHHFFF5AGAF2 NM:i:0 MD:Z:50 AS:i:50 XS:i:19 MQ:i:40 MC:Z:50M ms:i:1875
[186]:
!module load bedtools seqtk; \
bedtools intersect -a test.valid_pairs.rmdup.uq.st.bam -b ctcfpos.bed | samtools view | cut -f 1 > read.list ;\
seqtk subseq test.10K.R1.fastq.gz read.list
@M04990:66:000000000-G83PY:1:2104:3420:8655 1:N:0:TCGGACGATCATGGGTAGACGGACAAGTATGCAGCGCGCTCAAGCACGTGGATTACCGAGCAGTCGTACGCCGATGCGAAACATCGGCCACTCTACGAC+TAGATCGC
GGCTGGAGTGCAGTGGTGCGATCTCGGCTCACTGCAACCTCTGCCTCCCA
+
ABBBBBFFFFFFGGGGGFGFGGGHHHGGGGFHHBHHHHHHHHHHHHHHGH
@M04990:66:000000000-G83PY:1:1101:17665:21940 1:N:0:TCGGACGATCATGGGTGATACGTCAAGTATGCAGCGCGCTCAAGCACGTGGATTGTACCTTAGTCGTACGCCGATGCGAAACATCGGCCACTTCTGTGT+TAGATCGC
GTTCACGCCTCGCGTATAATGTCTCGCGACAAGGGCGTTTCACTAGCACG
+
CCCCBFBFCFCBGGGGGGGGGGHHGGFGGGGGHHGGGGGGFHHHHHHHHG
BED examples¶
bed overlaps using intervene¶
https://intervene.readthedocs.io/en/latest/index.html
[189]:
!ls bedtools_test/*bed
bedtools_test/B_cell.bed bedtools_test/HSPC_LOC.bed
bedtools_test/CD34_D0.bed bedtools_test/mono_cell.bed
bedtools_test/Ery_cell.bed bedtools_test/T_cell.bed
[203]:
!intervene upset --figtype png -i bedtools_test/*bed -o upset_plot --save-overlaps
Running UpSet module. Please wait...
You are done! Please check your results @ upset_plot.
Thank you for using Intervene!
[204]:
!ls upset_plot/* | head
upset_plot/Intervene_upset_combinations.txt
upset_plot/Intervene_upset.pdf
upset_plot/Intervene_upset.png
upset_plot/Intervene_upset.R
upset_plot/sets:
000001_T_cell.bed
000010_mono_cell.bed
000011_mono_cell_T_cell.bed
001000_Ery_cell.bed
[205]:
from IPython.display import Image
Image("upset_plot/Intervene_upset.png")
[205]:
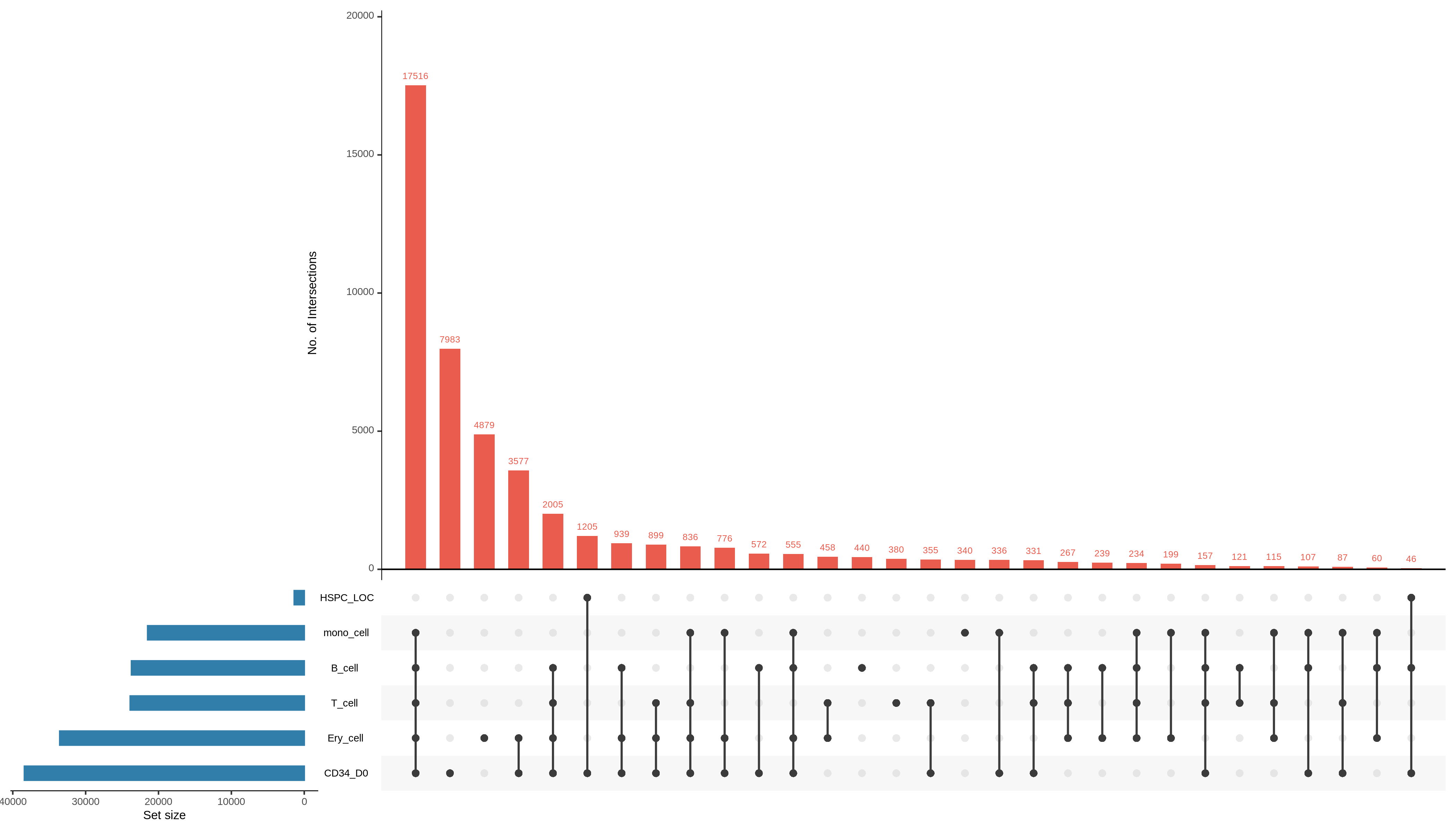
[206]:
!intervene pairwise --figtype png -i bedtools_test/*bed -o pairwise_frac_plot --compute frac --htype pie
Performing a pairwise intersection analysis. Please wait...
You are done! Please check your results @ pairwise_frac_plot.
Thank you for using Intervene!
[207]:
!ls pairwise_frac_plot/* | head
pairwise_frac_plot/Intervene_pairwise_frac_matrix.txt
pairwise_frac_plot/Intervene_pairwise_frac.png
pairwise_frac_plot/Intervene_pairwise_frac.R
[222]:
from IPython.display import Image
Image("pairwise_frac_plot/Intervene_pairwise_frac.png", width=500, height=500)
[222]:
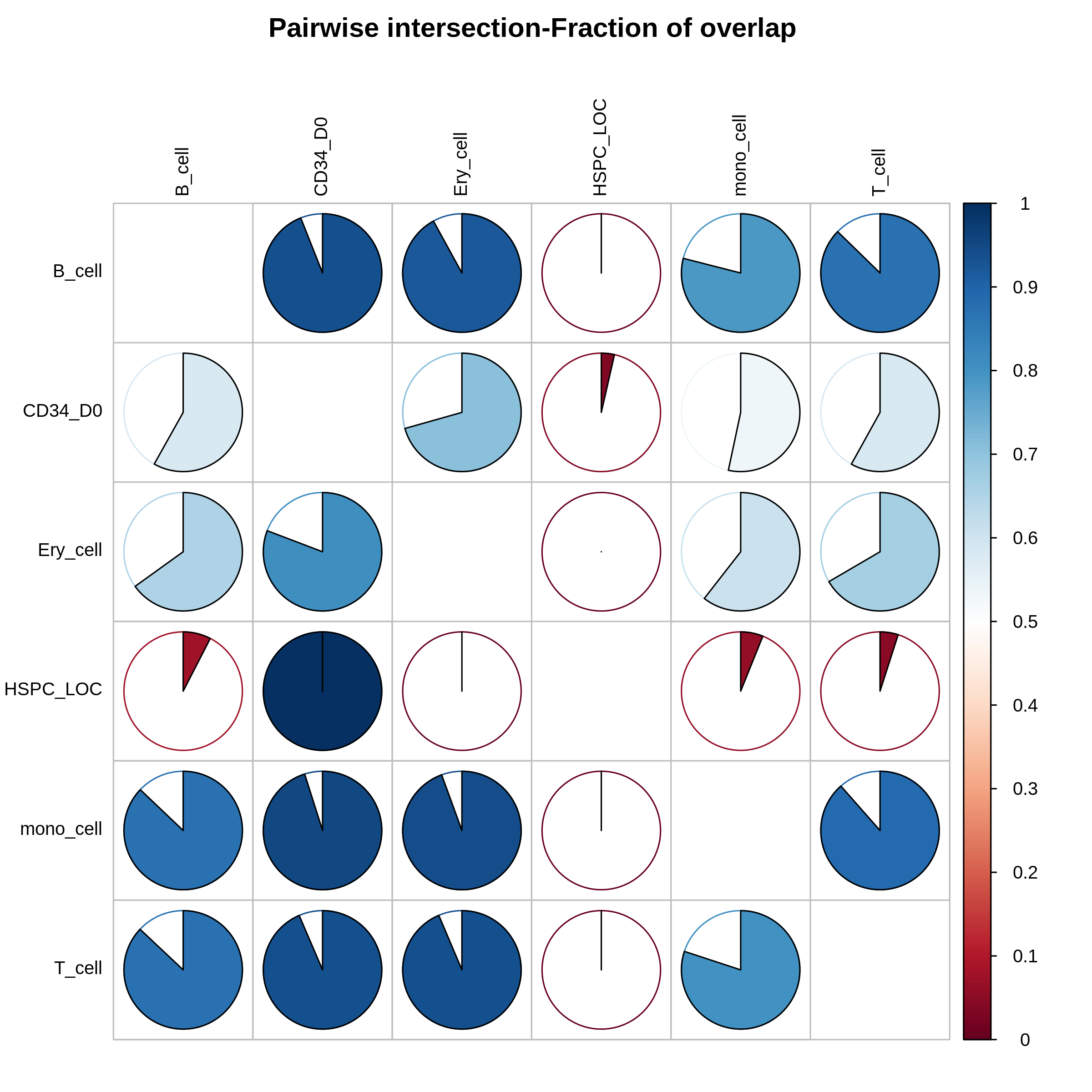
[209]:
!intervene venn --figtype png -i bedtools_test/*bed -o venn_plot --save-overlaps
Generating a 6-way "venn" diagram. Please wait...
Done! Please check your results @ venn_plot.
Thank you for using Intervene!
[210]:
!ls venn_plot/* | head
venn_plot/Intervene_venn.png
venn_plot/sets:
000001_T_cell.bed
000010_mono_cell.bed
000011_mono_cell_T_cell.bed
001000_Ery_cell.bed
001001_Ery_cell_T_cell.bed
001010_Ery_cell_mono_cell.bed
001011_Ery_cell_mono_cell_T_cell.bed
[220]:
from IPython.display import Image
Image("venn_plot/Intervene_venn.png", width=500, height=500)
[220]:
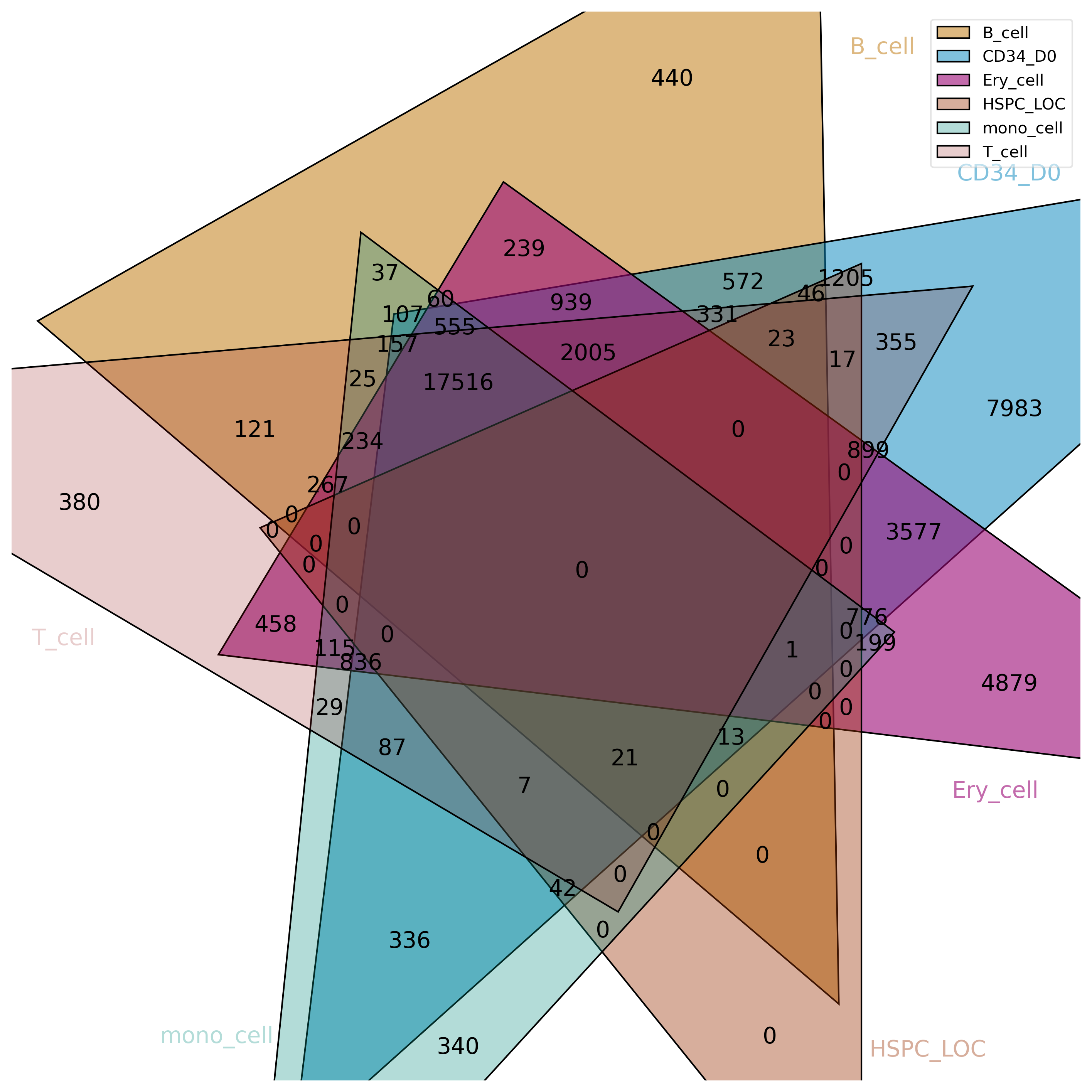
BEDTools¶
[223]:
!bedtools
bedtools is a powerful toolset for genome arithmetic.
Version: v2.30.0
About: developed in the quinlanlab.org and by many contributors worldwide.
Docs: http://bedtools.readthedocs.io/
Code: https://github.com/arq5x/bedtools2
Mail: https://groups.google.com/forum/#!forum/bedtools-discuss
Usage: bedtools <subcommand> [options]
The bedtools sub-commands include:
[ Genome arithmetic ]
intersect Find overlapping intervals in various ways.
window Find overlapping intervals within a window around an interval.
closest Find the closest, potentially non-overlapping interval.
coverage Compute the coverage over defined intervals.
map Apply a function to a column for each overlapping interval.
genomecov Compute the coverage over an entire genome.
merge Combine overlapping/nearby intervals into a single interval.
cluster Cluster (but don't merge) overlapping/nearby intervals.
complement Extract intervals _not_ represented by an interval file.
shift Adjust the position of intervals.
subtract Remove intervals based on overlaps b/w two files.
slop Adjust the size of intervals.
flank Create new intervals from the flanks of existing intervals.
sort Order the intervals in a file.
random Generate random intervals in a genome.
shuffle Randomly redistribute intervals in a genome.
sample Sample random records from file using reservoir sampling.
spacing Report the gap lengths between intervals in a file.
annotate Annotate coverage of features from multiple files.
[ Multi-way file comparisons ]
multiinter Identifies common intervals among multiple interval files.
unionbedg Combines coverage intervals from multiple BEDGRAPH files.
[ Paired-end manipulation ]
pairtobed Find pairs that overlap intervals in various ways.
pairtopair Find pairs that overlap other pairs in various ways.
[ Format conversion ]
bamtobed Convert BAM alignments to BED (& other) formats.
bedtobam Convert intervals to BAM records.
bamtofastq Convert BAM records to FASTQ records.
bedpetobam Convert BEDPE intervals to BAM records.
bed12tobed6 Breaks BED12 intervals into discrete BED6 intervals.
[ Fasta manipulation ]
getfasta Use intervals to extract sequences from a FASTA file.
maskfasta Use intervals to mask sequences from a FASTA file.
nuc Profile the nucleotide content of intervals in a FASTA file.
[ BAM focused tools ]
multicov Counts coverage from multiple BAMs at specific intervals.
tag Tag BAM alignments based on overlaps with interval files.
[ Statistical relationships ]
jaccard Calculate the Jaccard statistic b/w two sets of intervals.
reldist Calculate the distribution of relative distances b/w two files.
fisher Calculate Fisher statistic b/w two feature files.
[ Miscellaneous tools ]
overlap Computes the amount of overlap from two intervals.
igv Create an IGV snapshot batch script.
links Create a HTML page of links to UCSC locations.
makewindows Make interval "windows" across a genome.
groupby Group by common cols. & summarize oth. cols. (~ SQL "groupBy")
expand Replicate lines based on lists of values in columns.
split Split a file into multiple files with equal records or base pairs.
summary Statistical summary of intervals in a file.
[ General Parameters ]
--cram-ref Reference used by a CRAM input
[ General help ]
--help Print this help menu.
--version What version of bedtools are you using?.
--contact Feature requests, bugs, mailing lists, etc.
Most common bedtools commands¶
intersect, Find overlapping intervals in various ways.
closest, Find the closest, potentially non-overlapping interval.
merge, Combine overlapping/nearby intervals into a single interval.
slop, Adjust the size of intervals.
sort , Order the intervals in a file.
Other subcommand usage is very similar, you can read invidual documentation for more details. Just google bedtools
bedtools intersect¶
[226]:
!ls hg19-blacklist.v2.bed.gz
hg19-blacklist.v2.bed.gz
[227]:
!ls *bed
bd9835eb2b5b.bed example.bed matches.bed.bed
ca218748b8db.bed get_promoter_yli11_2019-12-10.bed out.bed
CTCF.hg38.random.bed get_promoter_yli11_2019-12-18.bed output.bed
ctcfneg_1x.bed get_promoter_yli11_2020-01-06.bed test23.bed
ctcfpos.bed input.bed test2.bed
ctcfpos.neg.bed input.bed.assigned_targets.bed test.bed
d3.bed loci.bed tmp.out.bed
df1.bed ma.bed
df2.bed matches.bed
[229]:
!bedtools intersect -a ctcfpos.neg.bed -b hg19-blacklist.v2.bed.gz | head
chr17 19033560 19033878
chr22 16741870 16742161
chr22 15446088 15446374
chr1 144198266 144198544
chr8 12379842 12380140
chr22 16266062 16266356
chr22 21843468 21843748
chr5 69578590 69578870
chrX 114989405 114989716
chr12 495349 495656
[230]:
!bedtools intersect -a ctcfpos.neg.bed -b hg19-blacklist.v2.bed.gz | bedtools sort -i - | head
chr1 121146526 121146821
chr1 142089885 142090188
chr1 144198266 144198544
chr11 1148412 1148715
chr12 495349 495656
chr14 18713048 18713369
chr14 19136813 19137201
chr15 19568430 19568738
chr15 19973033 19973368
chr15 21664313 21664604
[232]:
!bedtools intersect -b ctcfpos.neg.bed -a hg19-blacklist.v2.bed.gz -u | bedtools sort -i - | head
chr1 120926100 121149300 High Signal Region
chr1 121341500 145396500 High Signal Region
chr11 1141100 1214300 High Signal Region
chr12 479900 531700 High Signal Region
chr14 0 20303800 High Signal Region
chr15 0 20166200 High Signal Region
chr15 20200400 22365100 High Signal Region
chr15 30358500 30919300 High Signal Region
chr15 32445900 32915200 High Signal Region
chr15 82582300 83213900 High Signal Region
[233]:
!bedtools intersect -b ctcfpos.neg.bed -a hg19-blacklist.v2.bed.gz | bedtools sort -i - | head
chr1 121146526 121146821 High Signal Region
chr1 142089885 142090188 High Signal Region
chr1 144198266 144198544 High Signal Region
chr11 1148412 1148715 High Signal Region
chr12 495349 495656 High Signal Region
chr14 18713048 18713369 High Signal Region
chr14 19136813 19137201 High Signal Region
chr15 19568430 19568738 High Signal Region
chr15 19973033 19973368 High Signal Region
chr15 21664313 21664604 High Signal Region
remove black list regions¶
[234]:
!bedtools intersect -a ctcfpos.neg.bed -b hg19-blacklist.v2.bed.gz -v -wa | bedtools sort -i - | head
chr1 1261101 1261398
chr1 1729311 1729599
chr1 2245235 2245525
chr1 3153860 3154143
chr1 3244752 3245042
chr1 3418422 3418758
chr1 4443875 4444191
chr1 5243988 5244261
chr1 5641142 5641440
chr1 6010760 6011094
[237]:
!bedtools intersect -a ctcfpos.neg.bed -b hg19-blacklist.v2.bed.gz -wa -wb | bedtools sort -i - | head
chr1 121146526 121146821 chr1 120926100 121149300 High Signal Region
chr1 142089885 142090188 chr1 121341500 145396500 High Signal Region
chr1 144198266 144198544 chr1 121341500 145396500 High Signal Region
chr11 1148412 1148715 chr11 1141100 1214300 High Signal Region
chr12 495349 495656 chr12 479900 531700 High Signal Region
chr14 18713048 18713369 chr14 0 20303800 High Signal Region
chr14 19136813 19137201 chr14 0 20303800 High Signal Region
chr15 19568430 19568738 chr15 0 20166200 High Signal Region
chr15 19973033 19973368 chr15 0 20166200 High Signal Region
chr15 21664313 21664604 chr15 20200400 22365100 High Signal Region
[246]:
!bedtools intersect -b ctcfpos.neg.bed -a hg19-blacklist.v2.bed.gz -wa
chr11 1141100 1214300 High Signal Region
chr12 479900 531700 High Signal Region
chr14 0 20303800 High Signal Region
chr14 0 20303800 High Signal Region
chr15 0 20166200 High Signal Region
chr15 0 20166200 High Signal Region
chr15 20200400 22365100 High Signal Region
chr15 20200400 22365100 High Signal Region
chr15 30358500 30919300 High Signal Region
chr15 32445900 32915200 High Signal Region
chr15 82582300 83213900 High Signal Region
chr15 82582300 83213900 High Signal Region
chr17 4734800 4736700 Low Mappability
chr17 18928600 19140800 High Signal Region
chr17 22207000 25341300 High Signal Region
chr17 22207000 25341300 High Signal Region
chr18 15293900 18552900 High Signal Region
chr1 120926100 121149300 High Signal Region
chr1 121341500 145396500 High Signal Region
chr1 121341500 145396500 High Signal Region
chr21 9594900 10366000 High Signal Region
chr22 0 16962100 High Signal Region
chr22 0 16962100 High Signal Region
chr22 0 16962100 High Signal Region
chr22 20304800 20708400 High Signal Region
chr22 21466100 21916600 High Signal Region
chr22 21466100 21916600 High Signal Region
chr2 87441300 88290400 High Signal Region
chr2 90267000 95326200 High Signal Region
chr2 90267000 95326200 High Signal Region
chr2 97718400 98232300 High Signal Region
chr3 75678100 75917700 High Signal Region
chr3 197798000 198022400 High Signal Region
chr4 190795300 191154200 High Signal Region
chr5 68830000 70669400 High Signal Region
chr5 68830000 70669400 High Signal Region
chr6 62371500 62383900 Low Mappability
chr8 12252000 12466300 High Signal Region
chrX 114959100 115006100 High Signal Region
chrY 7432700 13491000 High Signal Region
[247]:
!bedtools intersect -b ctcfpos.neg.bed -a hg19-blacklist.v2.bed.gz -wa | bedtools sort -i - | bedtools merge -i - -o count -c 4
chr1 120926100 121149300 1
chr1 121341500 145396500 2
chr11 1141100 1214300 1
chr12 479900 531700 1
chr14 0 20303800 2
chr15 0 20166200 2
chr15 20200400 22365100 2
chr15 30358500 30919300 1
chr15 32445900 32915200 1
chr15 82582300 83213900 2
chr17 4734800 4736700 1
chr17 18928600 19140800 1
chr17 22207000 25341300 2
chr18 15293900 18552900 1
chr2 87441300 88290400 1
chr2 90267000 95326200 2
chr2 97718400 98232300 1
chr21 9594900 10366000 1
chr22 0 16962100 3
chr22 20304800 20708400 1
chr22 21466100 21916600 2
chr3 75678100 75917700 1
chr3 197798000 198022400 1
chr4 190795300 191154200 1
chr5 68830000 70669400 2
chr6 62371500 62383900 1
chr8 12252000 12466300 1
chrX 114959100 115006100 1
chrY 7432700 13491000 1
merge peaks¶
[249]:
!ls *bed
bd9835eb2b5b.bed example.bed matches.bed.bed
ca218748b8db.bed get_promoter_yli11_2019-12-10.bed out.bed
CTCF.hg38.random.bed get_promoter_yli11_2019-12-18.bed output.bed
ctcfneg_1x.bed get_promoter_yli11_2020-01-06.bed test23.bed
ctcfpos.bed input.bed test2.bed
ctcfpos.neg.bed input.bed.assigned_targets.bed test.bed
d3.bed loci.bed tmp.out.bed
df1.bed ma.bed
df2.bed matches.bed
[254]:
!cat ctcfpos.bed ctcfpos.neg.bed | bedtools sort -i - | bedtools merge -i - > merge.bed; head merge.bed
chr1 535747 536177
chr1 1261101 1261398
chr1 1430705 1431030
chr1 1729311 1729599
chr1 2245235 2245525
chr1 2303085 2303376
chr1 3153860 3154143
chr1 3244752 3245042
chr1 3418422 3418758
chr1 4443875 4444191
bedtools closest¶
[256]:
!bedtools sort -i ctcfpos.bed > t1.bed
!bedtools sort -i ctcfpos.neg.bed > t2.bed
!bedtools closest -a t1.bed -b t2.bed -d | head
chr1 535747 536177 chr1 1261101 1261398 724925
chr1 1430705 1431030 chr1 1261101 1261398 169308
chr1 2303085 2303376 chr1 2245235 2245525 57561
chr1 5492536 5492843 chr1 5641142 5641440 148300
chr1 6326110 6326418 chr1 6010760 6011094 315017
chr1 6702688 6702985 chr1 6010760 6011094 691595
chr1 8296980 8297279 chr1 8295680 8295961 1020
chr1 8831580 8831868 chr1 8878641 8878924 46774
chr1 8970722 8971025 chr1 8878641 8878924 91799
chr1 9093414 9093739 chr1 8878641 8878924 214491
extend bed file by certain size¶
[258]:
!bedtools slop -h
Tool: bedtools slop (aka slopBed)
Version: v2.30.0
Summary: Add requested base pairs of "slop" to each feature.
Usage: bedtools slop [OPTIONS] -i <bed/gff/vcf> -g <genome> [-b <int> or (-l and -r)]
Options:
-b Increase the BED/GFF/VCF entry -b base pairs in each direction.
- (Integer) or (Float, e.g. 0.1) if used with -pct.
-l The number of base pairs to subtract from the start coordinate.
- (Integer) or (Float, e.g. 0.1) if used with -pct.
-r The number of base pairs to add to the end coordinate.
- (Integer) or (Float, e.g. 0.1) if used with -pct.
-s Define -l and -r based on strand.
E.g. if used, -l 500 for a negative-stranded feature,
it will add 500 bp downstream. Default = false.
-pct Define -l and -r as a fraction of the feature's length.
E.g. if used on a 1000bp feature, -l 0.50,
will add 500 bp "upstream". Default = false.
-header Print the header from the input file prior to results.
Notes:
(1) Starts will be set to 0 if options would force it below 0.
(2) Ends will be set to the chromosome length if requested slop would
force it above the max chrom length.
(3) The genome file should tab delimited and structured as follows:
<chromName><TAB><chromSize>
For example, Human (hg19):
chr1 249250621
chr2 243199373
...
chr18_gl000207_random 4262
Tips:
One can use the UCSC Genome Browser's MySQL database to extract
chromosome sizes. For example, H. sapiens:
mysql --user=genome --host=genome-mysql.cse.ucsc.edu -A -e \
"select chrom, size from hg19.chromInfo" > hg19.genome
[259]:
!chrom_size=/home/yli11/Data/Human/hg19/annotations/hg19.chrom.sizes;\
bedtools slop -i ctcfpos.bed -g $chrom_size -b 10 | head
chr1 36659006 36659360
chr1 67738730 67739065
chr1 15630222 15630586
chr1 9850142 9850470
chr1 43767393 43767722
chr1 26616134 26616456
chr1 151462162 151462521
chr1 27243619 27243952
chr1 47431156 47431494
chr1 44243924 44244249
[260]:
!head ctcfpos.bed
chr1 36659016 36659350
chr1 67738740 67739055
chr1 15630232 15630576
chr1 9850152 9850460
chr1 43767403 43767712
chr1 26616144 26616446
chr1 151462172 151462511
chr1 27243629 27243942
chr1 47431166 47431484
chr1 44243934 44244239
[261]:
!chrom_size=/home/yli11/Data/Human/hg19/annotations/hg19.chrom.sizes;\
bedtools slop -i ctcfpos.bed -g $chrom_size -l 1 -r 10 | head
chr1 36659015 36659360
chr1 67738739 67739065
chr1 15630231 15630586
chr1 9850151 9850470
chr1 43767402 43767722
chr1 26616143 26616456
chr1 151462171 151462521
chr1 27243628 27243952
chr1 47431165 47431494
chr1 44243933 44244249
Bigwiggle Format¶
[ ]:
## Bigwiggle Format