Quantify prime editor off-target activity for crispressoPooled experiments¶
usage: crispressoPooled_PE.py [-h] [-j JID] -a AMPLICON_BED -gRNA GRNA_BED -f
FASTQ_TSV --RTT RTT [--remove_PAM]
[--queue QUEUE] [-g GENOME]
[--genome_fasta GENOME_FASTA]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
crispressoPooled_PE_yli11_2022-09-07)
-a AMPLICON_BED, --amplicon_bed AMPLICON_BED
amplicon_bed required, name column need to match gRNA
bed (default: None)
-gRNA GRNA_BED, --gRNA_bed GRNA_BED
gRNA_bed required, name column need to match amp bed
(default: None)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
gRNA_bed required (default: None)
--RTT RTT reverse transcription template. Note, this sequence is
used to check substitution rate caused by prime
editing, so for this parameter, you should use the
reverse complement sequence of the actual RTT sequence
in pegRNA (default: None)
--remove_PAM gRNA bed coord include PAM, but crispresso need to
remove PAM (default: False)
--queue QUEUE which queue to use (default: standard)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
--genome_fasta GENOME_FASTA
genome fasta file (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
Summary¶
Given off-target region, we quantify:
indel frequency. Any insertion or deletion that overlap with cas9 cutting position, exactly the same as in crisprEsso
--quantification_window_size 1 --quantification_window_center -3substitution rate. We quantify the first mismatch to the template sequence (i.e., expected mutated sequence). See the example below.
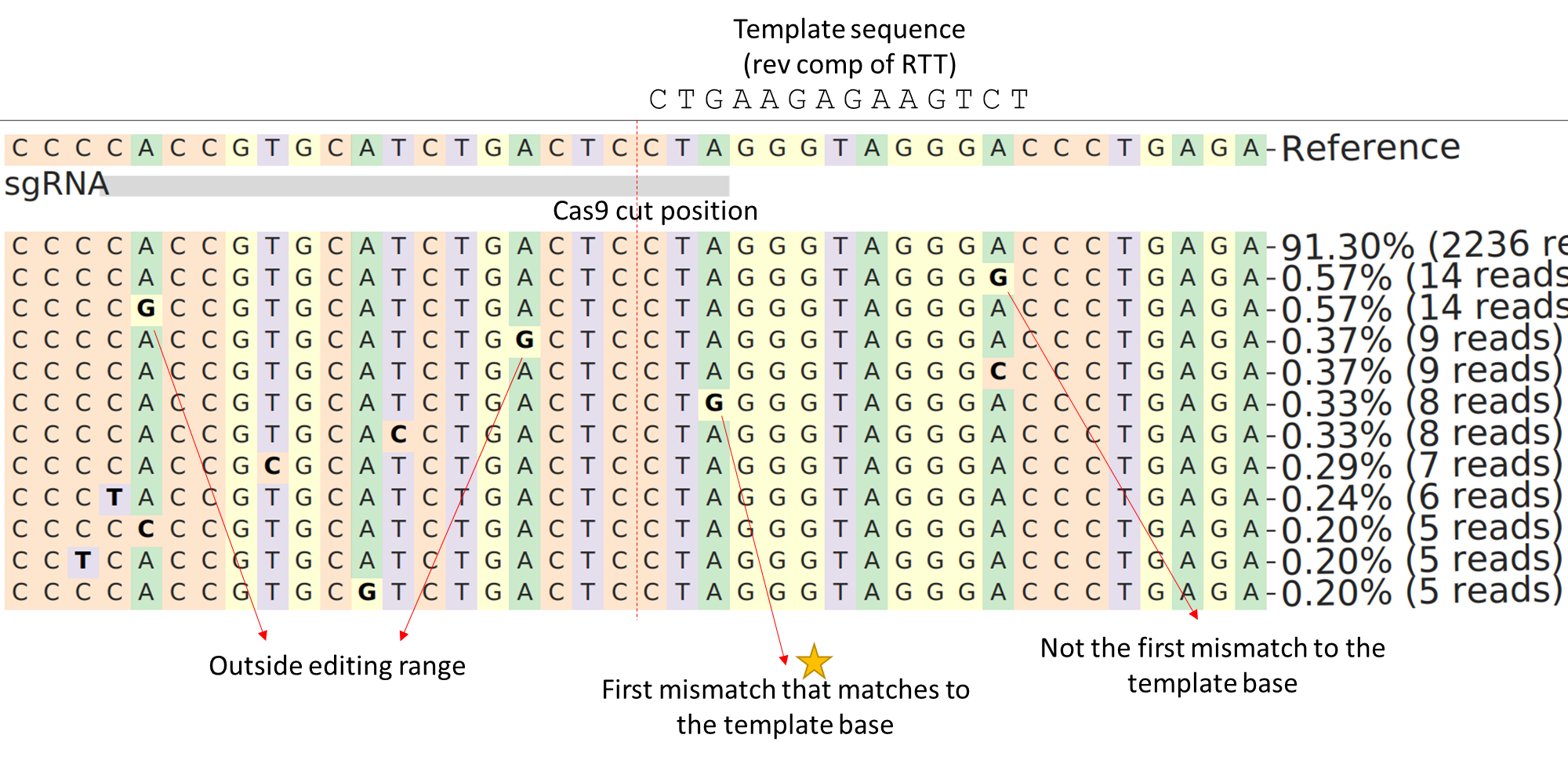
Input¶
The 4th column (name column) in the following two files should match (case sensitive!)
All these bed files should be bed6 format.
1. Amplicon sequence bed file¶
The amplicon sequence can be downloaded from IDT. Assay.bed
chr2 57769587 57769832 HBGg22_Target09 0 +
chrX 120721125 120721318 HBGg22_Target122 0 +
chr7 14859651 14859845 HBGg22_Target105 0 +
2. gRNA bed file¶
Header starting with “#” is acceptable.
#chr start end name CHANGEseq_reads strand
chr1 171455834 171455854 HBGg22_Target01 1 -
chr1 235564562 235564582 HBGg22_Target02 1 -
chr10 21466883 21466903 HBGg22_Target03 1 +
if the bed file coordinates include PAM, please use --remove_PAM option when submitting the job.
Output¶
In the jobID folder, you should be a summary stats csv file with columns showing the total reads and percentages of indel and subtitution. The percentage values are already multipled by 100.
sample site reads_total is_indel_total is_indel_percent is_sub_total is_sub_percent
xx A 39445 14 0.035492457852706 15 0.038027633413613406
xx B 61590 12 0.019483682415976103 10 0.0162364020133135
xx C 10409 2 0.0192141416082236 12 0.1152848496493417
Usage¶
Copy fastq files, amplicon bed file, and gRNA bed file in the working dir and run the following:
hpcf_interactive
export PATH=$PATH:"/home/yli11/HemTools/bin"
module load python/2.7.13
run_lsf.py --guess_input --single
crispressoPooled_PE.py -a 3AssayHBBALLOTs.bed -gRNA 3TargetHBBALLOTs.bed -f fastq.tsv --remove_PAM --RTT CTGAAGAGAAGTCT