Crispresso2 for Base editor¶
usage: crispresso2_BE.py [-h] [-j JID] [-r1 | -r2] [-f INPUT_LIST] [-q Q]
[--tsv TSV] [-s S] [-m MIN_OVERLAP] [--ref REF]
[--alt ALT]
[--base_editor_commands BASE_EDITOR_COMMANDS]
[--default] [--queue QUEUE]
[--gRNA_and_primers GRNA_AND_PRIMERS]
[--gRNA_and_primers_BWA GRNA_AND_PRIMERS_BWA]
[-g GENOME] [--genome_fasta GENOME_FASTA]
[--index_file INDEX_FILE]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
crispresso2_BE_yli11_2021-08-16)
-r1 only use R1 read for analysis (default: False)
-r2 only use R2 read for analysis (default: False)
-f INPUT_LIST, --input_list INPUT_LIST
tsv 5 columns, R1.fastq, R2.fastq, amplicon_seq,
gRNA_seq, output_name (default: None)
-q Q read quality (default: 0)
--tsv TSV not for end user (default: )
-s S base quality (default: 0)
-m MIN_OVERLAP, --min_overlap MIN_OVERLAP
min_overlap for read merging (default: 10)
--ref REF reference base (default: A)
--alt ALT alternative base (default: G)
--base_editor_commands BASE_EDITOR_COMMANDS
alternative base (default:
--quantification_window_size 10
--quantification_window_center -10
--base_editor_output --keep_intermediate --dump)
--default not running in base editor mode (default: False)
--queue QUEUE which queue to use (default: standard)
--gRNA_and_primers GRNA_AND_PRIMERS
tsv 4 columns, unique ID that matches to fastq file
name, gRNA seq, Forward Primer, Reverse Primer
(default: None)
--gRNA_and_primers_BWA GRNA_AND_PRIMERS_BWA
tsv 4 columns, unique ID that matches to fastq file
name, gRNA seq, Forward Primer, Reverse Primer
(default: None)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
--genome_fasta GENOME_FASTA
genome fasta file (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
--index_file INDEX_FILE
BWA index file (default: /home/yli11/Data/Human/hg19/i
ndex/bwa_16a_index/hg19.fa)
Note
Amplicon sequence and gRNA sequence should be upper case.
Summary¶
only work for hg19. But should be able to work on other genomes.
Running crispresso2 for base editor mode: https://github.com/pinellolab/CRISPResso2
The command is:
CRISPResso -r1 ${COL1}
-r2 ${COL2}
--amplicon_seq ${COL3}
--guide_seq ${COL4}
--quantification_window_size 10
--quantification_window_center -10
--base_editor_output -o {{jid}}/${COL5}
..note:: Note that the sgRNA sequence needs to be input as the guide RNA sequence (usually 20 nt) immediately adjacent to but not including the PAM sequence (5’ of NGG for SpCas9).
Updates¶
Users can now use -m to control minimal overlap for the FLASH command.
Added two more parameters for users to use only R1 or R2 for cirspresso analysis. For example:
To use only R1 read
crispresso2_BE.py -r1 --gRNA_and_primers input.list
To use only R2 read
crispresso2_BE.py -r2 --gRNA_and_primers input.list
Input¶
Option 1: User input amplicon sequence and gRNA sequence (-f)¶
A 5-column tsv file: R1.fastq, R2.fastq, amplicon_seq, gRNA_seq, output_name
12_S12_L001_R1_001.fastq.gz 12_S12_L001_R2_001.fastq.gz Amplicon_seq CTTGACCAATAGCCTTGACA test1
XXXX_L001_R1_001.fastq.gz XXXX_L001_R2_001.fastq.gz Amplicon_seq CTTGACCAATAGCCTTGACA Bababa
Option 2: User input Primer sequence and gRNA sequence (--gRNA_and_primers)¶
This option requires a unique ID in the fastq file name.
A 4-column tsv file: unique ID that matches to fastq file name, gRNA seq, Forward Primer, Reverse Primer
My fastq file is like:
gRNA1-ch19_S4_L001_R1_001.fastq.gz
gRNA2-ch19_S5_L001_R1_001.fastq.gz
gRNA5-ch2_S19_L001_R1_001.fastq.gz
gRNA8-ch8_S11_L001_R1_001.fastq.gz
gRNA1-ch19_S4_L001_R2_001.fastq.gz
gRNA2-ch19_S5_L001_R2_001.fastq.gz
gRNA5-ch2_S19_L001_R2_001.fastq.gz
gRNA10-ch8_S11_L001_R2_001.fastq.gz
Here, I can use gRNA1, gRNA2, etc. as my unique IDs. However, note that gRNA1 is a substring of gRNA10, so it is better to specify gRNA1- in your input file. So you will have something like:
gRNA1- gRNA_seq Forward_Primer Reverse_Primer
gRNA2 gRNA_seq Banana Orange
gRNA10 XXXXX BBBBBB AAAAAA
To get eff, use:
cd $jid
crispresso2_BE_get_eff.py ../input.list A G
Option 3: User input Primer sequence, gRNA sequence, and remove non-target matched reads (--gRNA_and_primers_BWA)¶
Everything is the same as option2, except that reads that mapped to other genomic regions will be removed from CrisprEsso2 analysis.
Only properly paired reads will be used. Duplicated reads are OK, non-uniquely mapped reads are OK if both R1 and R2 mapped to the target region (determined by in silico PCR).
Low-quality reads are filtered by CrisprEsso2: -q 10 -s 10, default is all 0.
Reads mapping quality is 40 and single base quality is Q40. See the table below.
Quality Score |
Error Probability |
Q40 |
0.0001 (1 in 10,000) |
Q30 |
0.001 (1 in 1,000) |
Q20 |
0.01 (1 in 100) |
Q10 |
0.1 (1 in 10) |
Usage¶
hpcf_interactive
module load python/2.7.13
crispresso2_BE.py -f input.list
OR
crispresso2_BE.py --gRNA_and_primers input.list
Output¶
Once the job is finished, you will receive a notification email.
Inside the jobID folder, you can look at the crispresso2 result. The html file is inside in each sub-folder.
crispresso2_BE.edit_eff.tsv This file contains the ref to alt base editing eff for position -15 to 20 (e.g., 0-20 is the gRNA sequence). The last column is the indel rate.
Usage2: Quantify custom mutations¶
This example is an improvement from previous example: custom mutation
Input <custom_mutation.tsv>¶
User needs to provide a 2-col tsv file, the first column is the reference sequence, the second column is the mutated sequence.
TCGATCACATTGT TCGATGCCATTGT
Command¶
Softlink your fastq files to a working dir:
hpcf_interactive
module load python/2.7.13
run_lsf.py --guess_input
amp=CTGTGGAAAATACCCAATTGCAGAACGAGAAACT # this is just an example, you should have longer sequence
gRNA=TCGATCACATTGT # gRNA sequences can be anything
crispresso2_BE.py -f fastq.tsv -a $amp -gRNA $gRNA --custom_mutation custom_mutation.tsv
Output¶
Here: edited_reads is the read counts for reference sequence. total_reads is the total number of aligned reads. editing_freq is the ratio.
label edited_reads total_reads editing_freq
pAC20_pool7_gRNA_sel_rep3_S21 117 188 0.622340426
pAC20_pool4_gRNA_sel_rep2_S11 185 1492 0.123994638
pAC21_pool5_gRNA_sel_rep2_S70 924 18761 0.049251106
pAC20_pool3_gRNA_sel_rep1_S7 61 494 0.123481781
pAC21_pool3_gRNA_nosel_rep1_S91 9188 33268 0.276181315
FAQ¶
Input format error¶
The input file requires specific number of columns. See some errors below:
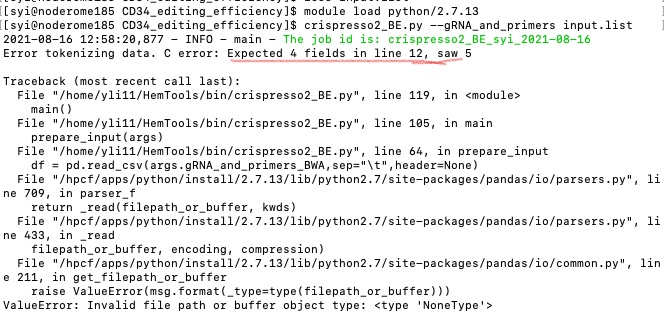
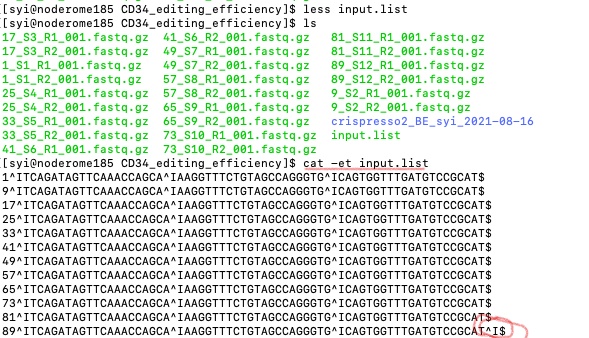
The solution to find these errors is using cat -et [your_file_name].
Video tutorial 1: a custom 2 gRNA base editing quantification¶
The command I actually used is: crispresso2_BE.py -f fastq.tsv --base_editor_commands " --quantification_window_size 30 --quantification_window_center 0 --base_editor_output --keep_intermediate --dump --plot_window_size 30" --interactive
Current HPC has a long waiting time, so I just run the pipeline interactively by adding --interactive. Also, I set the center to be somewhere in the middle of the two gRNA, --quantification_window_center 0 and increased the quantification window and visualization window size to 30bp.
Comments¶
code @ github.