sgRNA design for disrupting TFBS¶
usage: gRNA_design_TF.py [-h] [-j JID] -TAD TAD_FILE -peak PEAK_FILE -motif
MOTIF_FILE -gene GENE_FILE -off_target NUM_MATCH_FILE
[-l FLANKING_LENGTH] [--PAM_seq PAM_SEQ]
[--motif_position_anchor MOTIF_POSITION_ANCHOR]
[-g GENOME] [--genome_fasta GENOME_FASTA]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
gRNA_design_TF_yli11_2020-12-06)
-l FLANKING_LENGTH, --flanking_length FLANKING_LENGTH
number of bp flanking the motif bed file (default: 25)
--PAM_seq PAM_SEQ PAM seq (default: NGG)
--motif_position_anchor MOTIF_POSITION_ANCHOR
which position to use as +1 position, default is for
WGATAR, where the first A is used as +1 then T is -1
(default: 3)
required named arguments:
-TAD TAD_FILE, --TAD_file TAD_FILE
bed file, at least 4 columns (default: None)
-peak PEAK_FILE, --peak_file PEAK_FILE
bed file, at least 4 columns (default: None)
-motif MOTIF_FILE, --motif_file MOTIF_FILE
bed file, at 6 columns, containing strand (default:
None)
-gene GENE_FILE, --gene_file GENE_FILE
bed file, at least 4 columns (default: None)
-off_target NUM_MATCH_FILE, --Num_match_file NUM_MATCH_FILE
tsv file, 2 columns, sgRNA seq and number of matches
in the genome (default: None)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
chrom size. (default: hg19)
--genome_fasta GENOME_FASTA
genome version: hs, mm (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
Summary¶
This pipeline was developed in the GATA1 disruption project, but is generic for other sgRNA design projects focusing on TF motifs.
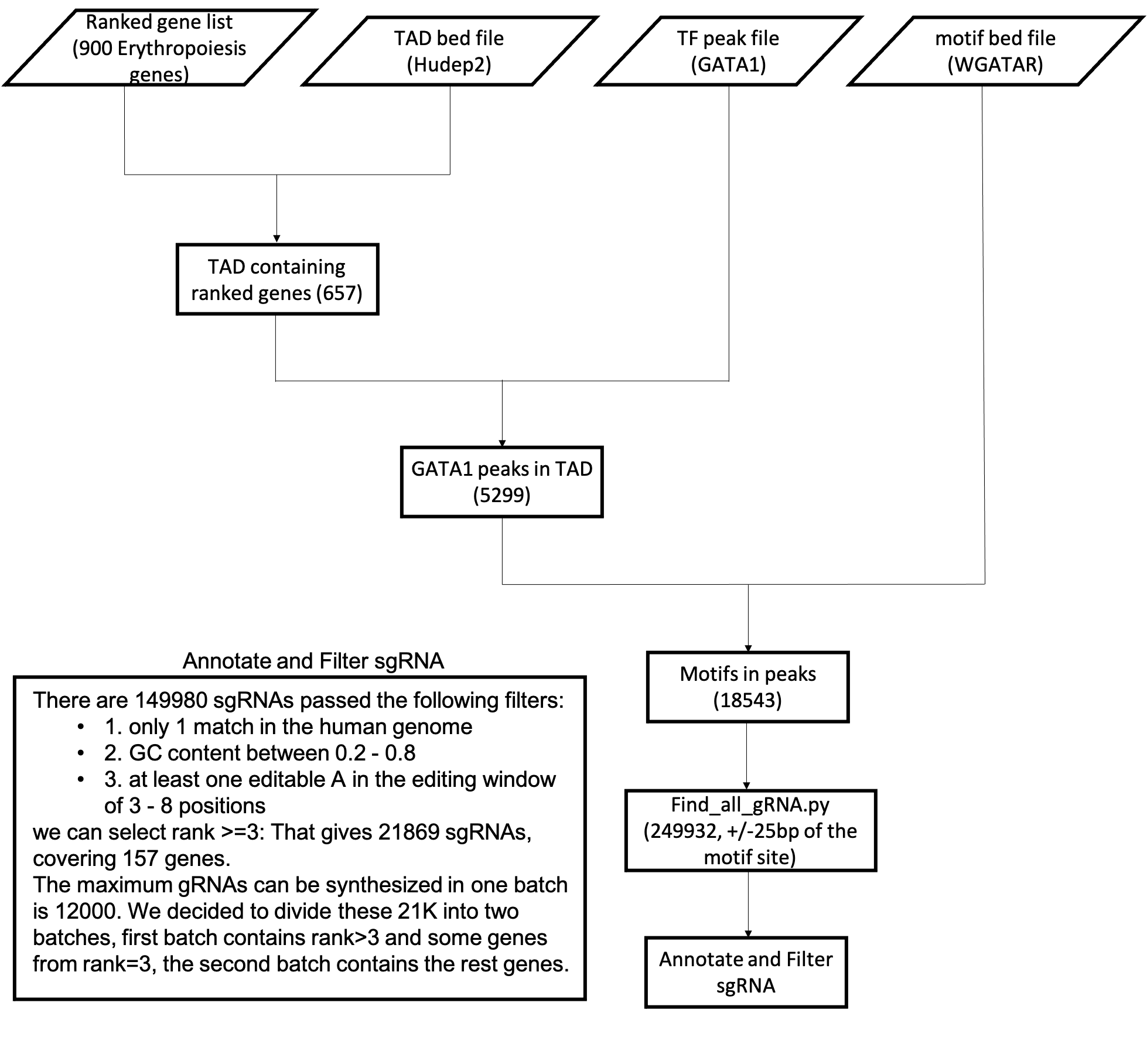
Input¶
You will need 4 bed files and 1 tsv file:
bed files for TF peaks, TF motifs, TADs, and gene bed file. Note that TF motif bed file requires 6 columns, where the last column is strand.
a tsv file for sgRNA genome-wide number of occurrences, first column is sgRNA_seq, second column is an integer.
Note
TAD.bed, gene.bed, peak.bed requires 4 columns, the last column is id or name. motif.bed requires 6 columns: chr, start, end, name, value, strand.
Usage¶
hpcf_interactive
module load python/2.7.13
gRNA_design_TF.py -TAD tad.bed -peak GATA1_peaks.bed -motif WGATAR.bed -gene ranked_genes.bed -off_target number_matches.bed -g hg19 --PAM NG -l 25 --motif_position_anchor 3
Output¶
The final output file is: Motif_{flanking_length}_gRNA.annot.bed.rel_edit_pos.filter.bed
It was filtered based on:
only 1 match in the genome
GC% : 0.2 - 0.8
at least one editable base in the editing window (predefined) 3-8
The output columns are:
0-5: sgRNA bed file
6-11: motif bed file
12-15: peak bed file
16: TAD name
17: gene name
18: relative position to the anchor ``--motif_position_anchor``
19: number of editable bases
20: number of off-targets, -1 means not found in the provided off-target table
Comments¶
code @ github.