Calculate custom defined editing frequency from Crispresso2 output¶
Summary¶
Specifically designed for calculating co-occurring mutations caused by 2 gRNAs (base editing).
But the function is generic.
Not work for counting indels.
Input¶
Allele frequency txt¶
Not really need to prepare this input after you finished running crispresso2_BE.py. I put it here for completeness because the program actually uses this file.
The output from crispresso, the visualization window (i.e., the actual sequence) must contain the two gRNA sequences, which means the default visualization parameters might not work.
mutation tsv¶
A 2-column tsv, the first column is the reference sequence (e.g., gRNA sequence), the second column is the mutated sequence (e.g., A-G conversion, the edited sequence). The reference sequence is not nessesarily the gRNA sequence, as long as we can uniquely locate the mutation (without doing reverse complement). For example, in the example below, we specified the reverse completement of the gRNA sequnece, so the mutation is actually T->C.
Another note: here I have two gRNAs in base editing and I want to check the co-occurring mutation. You are not required to specify two lines in mutation.tsv, you can just look at the allele frequency plot (the html file generated by crispresso2) and give the program a long DNA sequence that contains both mutations.
Usage¶
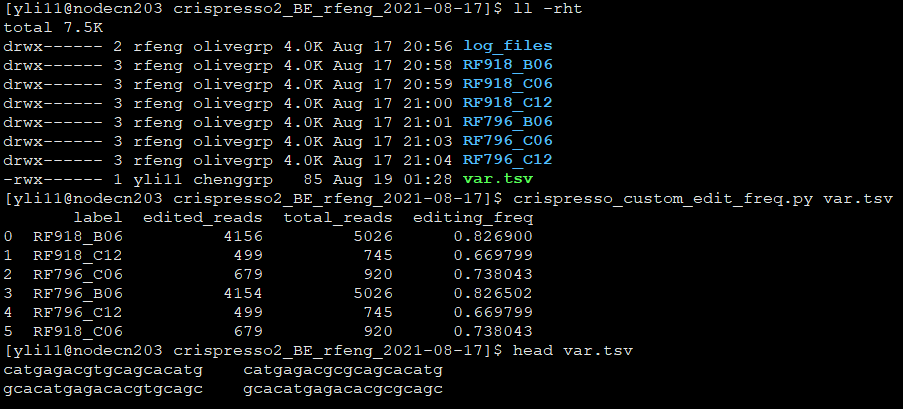
Go to the result folder containing all your crispresso2 outputs. For example, the jobID folder directly from running crispresso2_BE.py
hpcf_interactive
module load python/3.7.0
crispresso_custom_edit_freq.py var.tsv
Output¶
Output is saved as custom_edit_freq.csv.
Comments¶
code @ github.