subsample fastq and visualize in sequence logo¶
Summary¶
Mostly used for exploration and debug of raw fastq files, to identify common sequence patterns.
Steps: fastq was subsamples to 2M reads, clustered and reordered based on k-mer and sequence logo was generated for each of the 10-splits reads. For example, a sequence logo for 0% to 10% of the reads.
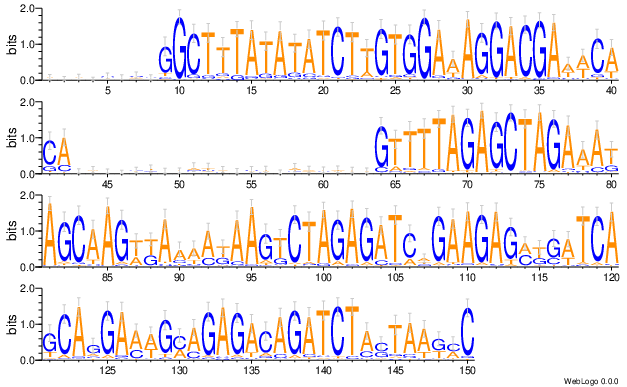
Usage¶
Copy your fastq to a working dir.
hpcf_interactive
module load python/2.7.13
run_lsf.py --guess_input --single
run_lsf.py -f fastq.tsv -p fastq_vis