Genomic features annotatoin given bed file¶
usage: annot_gene_features.py [-h] -f INPUT_BED [-g GENOME] [--tss TSS]
[--exon EXON] [--promoter PROMOTER]
[--UTR5 UTR5] [--UTR3 UTR3] [--intron INTRON]
[--gene_name_list GENE_NAME_LIST] [--coding]
[-d1 D1] [-d2 D2] [-o OUTPUT] [--gene_names]
optional arguments:
-h, --help show this help message and exit
-f INPUT_BED, --input_bed INPUT_BED
3 column bed file, additional columns are OK, but will
be ignored (default: None)
--coding focus on coding genes only (default: False)
-d1 D1 extend query bed for intersection (default: 0)
-d2 D2 extending genomic features for intersection (default:
0)
-o OUTPUT, --output OUTPUT
output intermediate file (default: output)
--gene_names use gene names instead of gene id, only works for hg19
now (default: False)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
the annotation file (default: hg19)
--tss TSS tss feature file, 4 columns, chr, start, end , gene
name (default: None)
--exon EXON exon feature file, 4 columns, chr, start, end , gene
name (default: None)
--promoter PROMOTER promoter feature file, 4 columns, chr, start, end ,
gene name (default: None)
--UTR5 UTR5 5UTR feature file, 4 columns, chr, start, end , gene
name (default: None)
--UTR3 UTR3 3UTR feature file, 4 columns, chr, start, end , gene
name (default: None)
--intron INTRON intron feature file, 4 columns, chr, start, end , gene
name (default: None)
--gene_name_list GENE_NAME_LIST
a file containing id to name conversion (default:
None)
Summary¶
Genomic features are based on Gencode annotation, which is then parsed to exon, promoter, 5UTR, 3UTR, intron, intergenic regions using : https://github.com/saketkc/gencode_regions
Feature assignment program is based on EPI assignment program
Input¶
Bed file with at least 3 columns: chr, start, end
Output¶
8 columns will be added to the input bed file:
The first 2 columns are nearest_TSS_gene, nearest_TSS_distance.
The next 5 columns are overlaps with exon_gene, promoter_gene, 5UTR_gene, 3UTR_gene, intron_gene.
The last columns is Genomic_features, the priority is Exon, Promoter, 5UTR, 3UTR, Intron, Intergenic.
By default, output uses gene Ensemble ID. You can use --gene_names option to output just gene names.
Usage¶
export PATH=$PATH:"/home/yli11/HemTools/bin"
hpcf_interactive.sh
module load conda3
source activate /home/yli11/.conda/envs/py2
annot_gene_features.py -f input.bed -o output.bed
# OR
annot_gene_features.py -f input.bed -o output.bed --gene_names
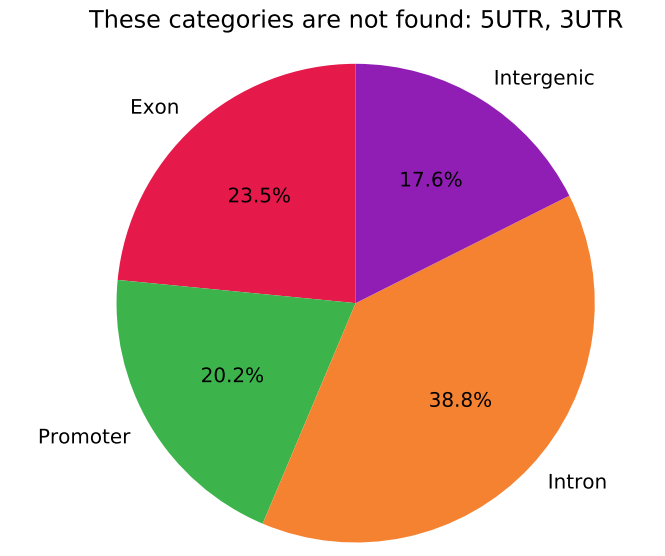
## generate a pie chart
pie_plot.py -f output.bed --order Exon,Promoter,5UTR,3UTR,Intron,Intergenic --use_col -1 --header
Pie chart example ^^^
n=3995
f=CFUe.06.peaks.bed
annotatePeaks.pl $f mm9 -gtf $ann > $f.annot.txt
pie_plot.py -f $f.annot.txt --use_col Annotation --header --homer -t "$f(n=$n)" -o $f.annot.pdf