Optimal subset finding problem in mutagenesis studies¶
usage: optimal_subset.py [-h] -f MUTATION_LIST [-dir OUTPUT_DIR] -a
AMPLICON_NUMBER [-t FLEXIBILITY] [-n TOP_N]
[-m MIN_SAMPLE]
[--binary_matrix_dir BINARY_MATRIX_DIR]
optional arguments:
-h, --help show this help message and exit
-f MUTATION_LIST, --mutation_list MUTATION_LIST
mutation list, can be a file with one mutation per
line or a list of mutations separated by comma
(default: None)
-dir OUTPUT_DIR, --output_dir OUTPUT_DIR
output_label (default:
optimal_subset_yli11_2020-05-06)
-a AMPLICON_NUMBER, --amplicon_number AMPLICON_NUMBER
amplicon number (default: None)
-t FLEXIBILITY, --flexibility FLEXIBILITY
+- t search space (default: 3)
-n TOP_N, --top_n TOP_N
save results for top n combinations for each t
(default: 5)
-m MIN_SAMPLE, --min_sample MIN_SAMPLE
if less than min_sample samples contain the given
combination, skip plot (default: 5)
--binary_matrix_dir BINARY_MATRIX_DIR
data matrix in feather format (default: /research/rgs0
1/project_space/tsaigrp/Genomics/common/projects/Cas9m
utagenesis/step1_mutation_count_yli11_2020-04-09/read_
count_matrix)
Summary¶
Finding a preferred subset from a given set of features is a common problem in biology. With different constraints, the problem can be mapped to set cover problem if user requires a minimal cardinality subset or mapped to feature selection problem if user wants to find the most distriminative subset between positive class and negative class. While the significance of individual feature can be quantified easily using statistical tests (e.g., DESEQ), the combination of features can’t be evaluated over the entire space because of the curse of dimensionality. One common approach to reduce the computational cost is to solve the problem through some kind of heuristic algorithm.
Here, we consider an optimal subset finding problem where the user has 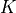 preferred features. The user would like to extend the features and find a superset that is of interest in terms of some metric. On the other direction, the user is also flexible on the “preferred” features; namely, the user would like to remove some feature(s) and find a subset that is of interest. In this study, we formally define the optimal subset finding problem and solve the problem via a greedy algorithm that finds sub-optimal solutions and a branch-and-bound algorithm that finds the exact optimal solution. We demonstrate the effectiveness of the solutions in mutagenesis studies.
preferred features. The user would like to extend the features and find a superset that is of interest in terms of some metric. On the other direction, the user is also flexible on the “preferred” features; namely, the user would like to remove some feature(s) and find a subset that is of interest. In this study, we formally define the optimal subset finding problem and solve the problem via a greedy algorithm that finds sub-optimal solutions and a branch-and-bound algorithm that finds the exact optimal solution. We demonstrate the effectiveness of the solutions in mutagenesis studies.
Todo
branch-and-bound algorithm
Problem definition¶
We consider a mutagenesis study where 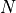 number of experiments have been sequenced and the total number of observed mutations is denoted by set
number of experiments have been sequenced and the total number of observed mutations is denoted by set 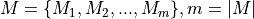 . The number of reads in each experiments is denoted by
. The number of reads in each experiments is denoted by 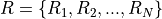 .
.
Given $k$ number of candidate mutations (e.g., selected for DESEQ2 results), the biological question is to examine if the co-occurring of the $k$ mutations, and the subset of the $k$ mutations or the superset of the $k$ mutations are also significantly enriched in each sample. In other words, we ask if adding $t$ new mutations to the set or removing $t$ mutations ($t<k$) from the set can produce more biological meaningful mutation sets.
A formal statement of this optimization problem is as follows. Let 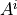 be a read count table for mutagenesis experiment $i$, $m$ be the number of observed mutations, and $r$ be the number of reads in sample $i$, where
be a read count table for mutagenesis experiment $i$, $m$ be the number of observed mutations, and $r$ be the number of reads in sample $i$, where
Input¶
1. mutation list¶
mutation_name1
mutation_name2
mutation_name3
Output¶
All output files are stored in the {{jid}} folder.
1. Summary.tsv¶
This file contains the number of reads, percentage of reads, D_score, and OE_score information for each mutation combination and each sample. I found OE_score is not informative.
When D_score>0, it represents the percentage of dropped reads when t mutations are added.
When D_score<0, its absolute value represents the presentage of increased reads when t mutations are removed.
mutation |
sample |
read_count |
D_score |
percent |
OE_score |
cardinality |
|---|---|---|---|---|---|---|
[‘x’, ‘mutation2’, ‘mutation3’] |
0-none.rep1.2 |
64 |
0.0 |
0.000382866714525006 |
3339.025469100123 |
3 |
[‘x’, ‘mutation2’] |
0-none.rep1.2 |
126 |
-0.96875 |
0.0007537688442211055 |
19.898871946034802 |
2 |
[‘mutation2’, ‘mutation3’] |
0-none.rep1.2 |
89 |
-0.390625 |
0.0005324240248863366 |
33.22221477858913 |
2 |
[‘x’, ‘mutation3’] |
0-none.rep1.2 |
65 |
-0.015625 |
0.0003888490069394592 |
17.954115827461766 |
2 |
[‘x’] |
0-none.rep1.2 |
1196 |
-17.6875 |
0.007154821727686048 |
1.0 |
1 |
[‘mutation2’] |
0-none.rep1.2 |
885 |
-12.828125 |
0.005294328786791098 |
1.0 |
1 |
[‘mutation3’] |
0-none.rep1.2 |
506 |
-6.90625 |
0.0030270399617133284 |
1.0 |
1 |
[‘x’, ‘mutation2’, ‘mutation3’] |
0-none.rep2.2 |
41 |
0.0 |
0.00046273305945555507 |
2489.549954985795 |
3 |
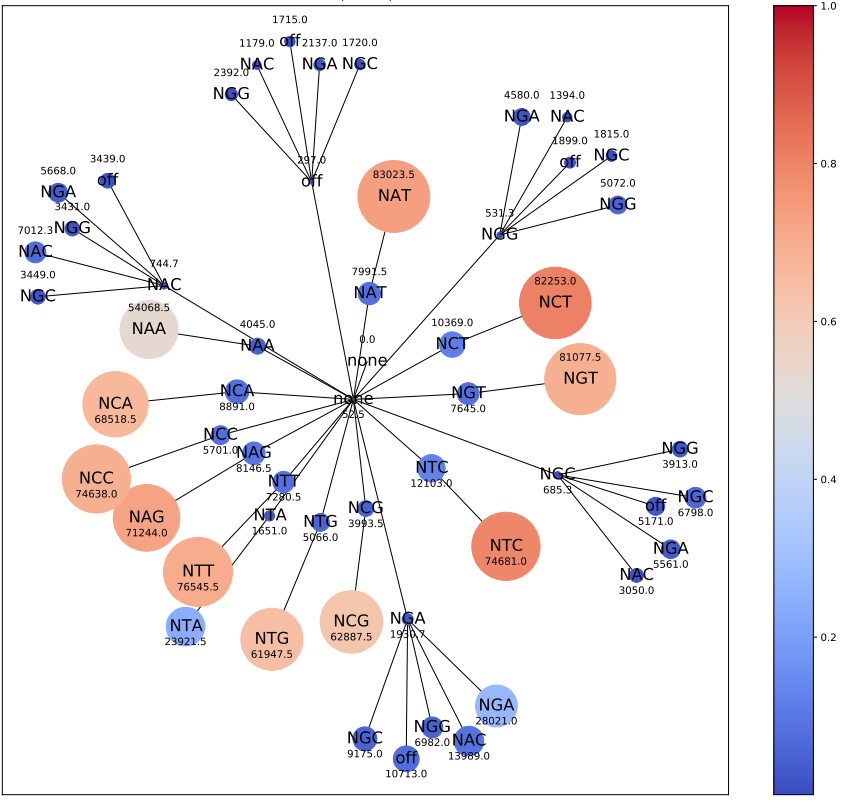
2. enrichment visualization¶
Node color represents percent of reads. Node size represent number of reads.
Figure title contains the mutation names (hidden for this example).
Usage¶
hpcf_interactive_large.sh
module load conda3
source activate /home/yli11/.conda/envs/py2
export PATH=$PATH:"/home/yli11/Tools/optimal_subset"
optimal_subset.py -f test_mutations.list -a 2
Notes on association rule mining¶
https://en.wikipedia.org/wiki/Association_rule_learning
https://github.com/chuanconggao/PrefixSpan-py
https://github.com/ChongYou/subspace-clustering
http://rasbt.github.io/mlxtend/user_guide/frequent_patterns/association_rules/