Homer ChIP-seq analysis¶
Reference: Homer NGS annotation
hpcf_interactive -q standard -R "rusage[mem=20000]"
module load homer/4.10
Peak annotation with genomic features: TSS, intron, exon, etc.¶
Using default genomes
annotatePeaks.pl [peak file] [genome version] -annStats annotate.log > output.tsv
[peak file] : narrowPeak file from HemTools
[genome version] : hg18, hg19, mm9, mm10.
output.tsv is the peak annotation file, such as TSS-promoter, exon, etc.
Find motifs¶
findMotifsGenome.pl [peak_file] [genome_version] myOutput -size 200 -mask -preparsedDir parsing_genome_dir
Ref: http://homer.ucsd.edu/homer/ngs/peakMotifs.html
Tip
Need to add “-preparsedDir parsing_genome_dir” homer will need to write a temp background files, you need some where that is writable.
Motif scanning: Find which peaks contain the motif¶
Different motif scanning tools will give similar but still different results. For consistency, if you used homer for motif discovery, you should use homer for motif scanning.
Input motif is homer motif format¶
>CCACHAGGKGGC 1-CCACHAGGKGGC,BestGuess:BORIS(Zf)/K562-CTCFL-ChIP-Seq(GSE32465)/Homer(0.929) 6.908069 -165.654312 0 T:203
.0(53.70%),B:7014.9(14.16%),P:1e-71 0.091 0.762 0.045 0.102 0.037 0.936 0.026 0.001 0.806 0.026 0.149 0.019 0.026 0.600 0.357 0.017 0.274 0.332 0.033 0.360 0.814 0.026 0.031 0.129 0.022 0.001 0.959 0.018 0.342 0.043 0.573 0.042 0.049 0.026 0.486 0.439 0.023 0.001 0.959 0.017 0.068 0.028 0.889 0.015 0.037 0.793 0.051 0.119
Note that 1-CCACHAGGKGGC,BestGuess:BORIS(Zf)/K562-CTCFL-ChIP-Seq(GSE32465)/Homer(0.929), this long name will be the motif name that homer uses.
You can find the motif PWM from the motif report.
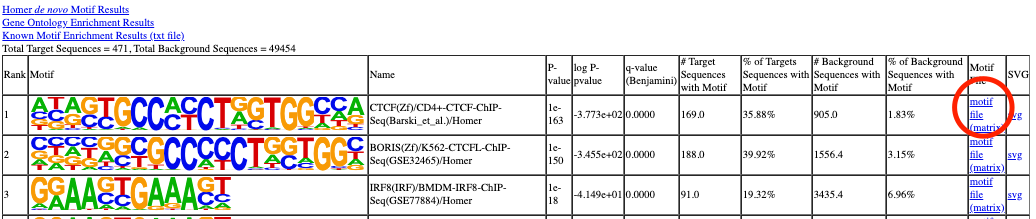
Usage¶
annotatePeaks.pl peak_file hg19 -m homer.motif > output.tsv
If you have N motifs in homer.motif, then the last N columns provide which peak contain the motif.
For example:
CTCF_B_cell_GCCCCCTRGTGG,BestGuess:BORIS(Zf)/K562-CTCFL-ChIP-Seq(GSE32465)/Homer(0.878) Distance From Peak(sequence,strand,conservation)
179(ACCCCCTGGCGG,+,0.00)
NA
NA means that the peak doesn’t contain the motif.
Motif co-occurrence in peaks¶
annotatePeaks.pl [peak file] [genome version] -annStats annotate.log -m [knownResults/*.motif] -matrix co_occur_motifs > output.tsv
[peak file] : narrowPeak file from HemTools
[genome version] : hg18, hg19, mm9, mm10.
[knownResults/*.motif] : findMotifsGenome.pl output dir.
Tip
You can also use -m /hpcf/apps/homer/install/4.9.1/motifs/*.motif. This is local homer motif database.
co_occur_motifs.stats.txt contains the co-occuring statistics.
output.tsv is the peak annotation file, with additional motif occurrence information.
Tip
You can control the peak size from the peak mid-point and use it to look for co-occuring motifs. For example, -size -300,300 will extend the peak to -300bp upstream from center and 300bp downstream.
annotatePeaks.pl [peak file] [genome version] -annStats annotate.log -m [knownResults/*.motif] -matrix co_occur_motifs -size -300,300 > output.tsv
[peak file]: for this size option, you might want to use the summits.bed file from HemTools.