Convert BCL basecall files to FASTQ files¶
Tip
For demultiplexing, you must provide SampleSheet.csv; otherwise, all fastq.gz files will be named as Undetermined. You have to upload SampleSheet.csv to the dir that conatins BCL files on HPC. Below is what this dir should look like (and the files and subfolders it should contain, not including fastq_files folder):
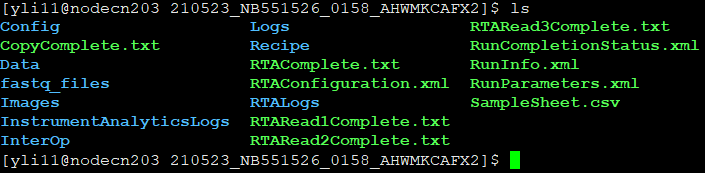
Note
SampleSheet.csv is a keyword, user much provide a file with exactly this name. Otherwise, the output will just be Undetermined. A different name is OK for more advanced users.
See this for SampleSheet format details: https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/sequencing-sheet-format-specifications-technical-note-970-2017-004.pdf
You can download an example SampleSheet.csv here: https://github.com/YichaoOU/HemTools/tree/master/docs/gallery/SampleSheet.csv
Note
Please note that there should be no spaces (i.e., only _ is allowed, for special char) in sample_ID (e.g., see some incorrect examples below). A unique sample_ID is also preferred.
Note
We found that for Miniseq and Nextseq, your R2 index in samplesheet.csv have to be reverse complemented. For Miseq, everything is normal.
Note
Basic sequence information is in RunInfo.xml if you are not sure about SE or PE or read/index length.
Step 1
hpcf_interactive -q standard -R "rusage[mem=32000]"
Step 2
module load bcl2fastq
Step 3
bcl2fastq --no-lane-splitting -o fastq_files
Once finished, you should be able to see the fastq files in folder fastq_files.
Note
fastq_files is the output folder. Feel free to change it.
Tip
By default, one mismatch is allowed for demultiplexing. If you want to allow for two mismatches, type the command below:
bcl2fastq --no-lane-splitting -o fastq_files --barcode-mismatches 2
Manually reverse complement R2 index¶
In some sequencer, e.g., miniseq or Nextseq, users have to manually get revcomp seq of R2 index. So in this case, you can use http://arep.med.harvard.edu/labgc/adnan/projects/Utilities/revcomp.html to get a list of revcomp sequences and copy and replace old index. Upload new sample sheet csv to HPC and run:
bcl2fastq --sample-sheet SampleSheet2.csv --no-lane-splitting -o fastq_files2
Wierd special char in your samplesheet.csv¶
If you have this Could not parse the CSV stream error, then likely you have some wierd special strings in your SampleSheet.csv.
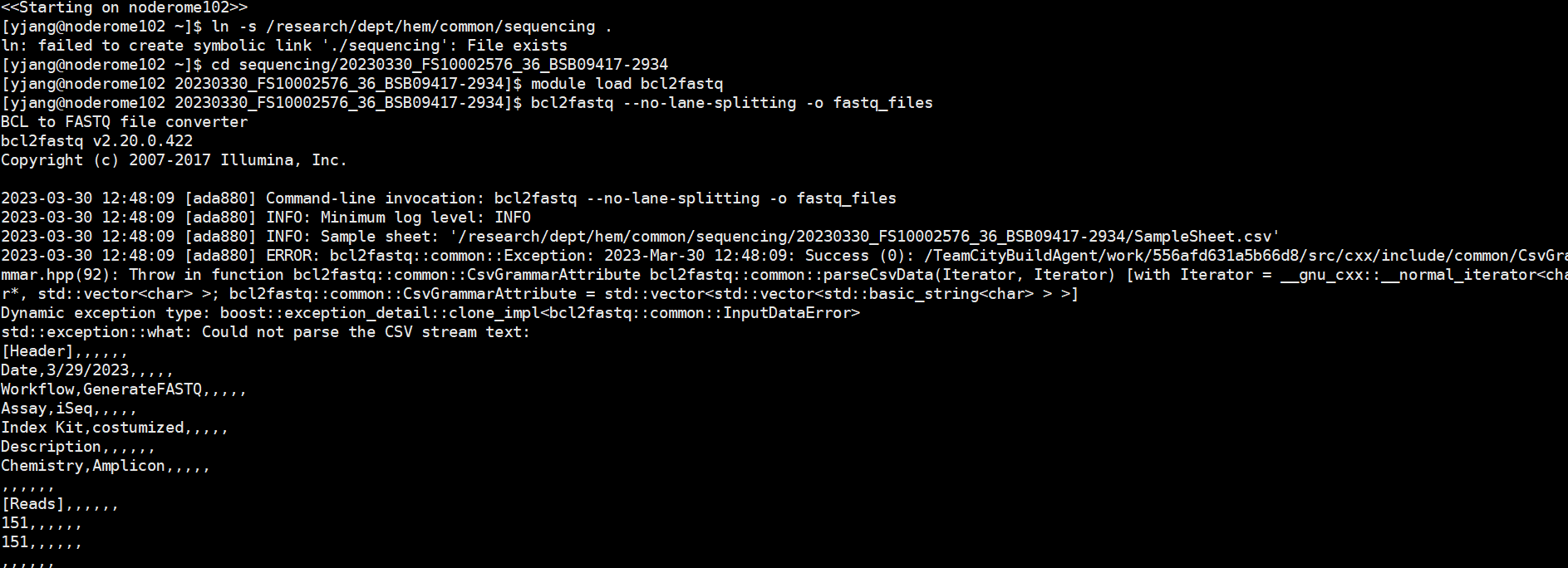
I have seem 3 types of specical strings. You can use less or cat -et to reveal these hidden strings.
<U+FEFF> character showing up in files. How to remove them?¶
https://gist.github.com/szydan/b225749445b3602083ed
1) In your terminal, open the file using vim:
vim file_name
2) Remove all BOM characters:
:set nobomb
3) Save the file:
:wq
<EF BB BF> character showing up in files. How to remove them?¶
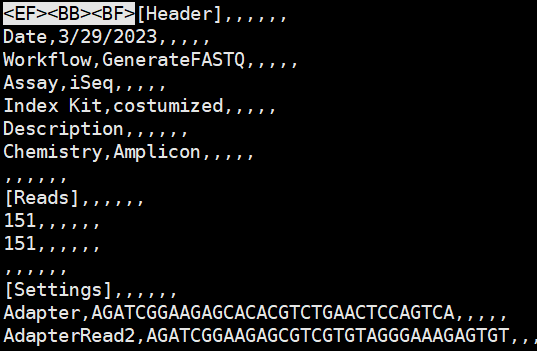
Just do this sed -i '1s/^\xEF\xBB\xBF//' SampleSheet.csv
Mising bcl¶
Question: “I have a pair-end 150bp sequencing run that stopped in the middle at about 150cycles. I think that it should contain the data that I need for the analysis. But when I do bcl files to fastq files, it showed error “Unable to find BCL file for ‘s_1_1102’ in”
Solution: Add --ignore-missing-bcls option.
For example: bcl2fastq --no-lane-splitting -o fastq_files --ignore-missing-bcls
Which delmultiplexing pipeline I should use¶
Generating I1 I2 reads¶
bcl2fastq --no-lane-splitting -o fastq_files2 --create-fastq-for-index-reads --ignore-missing-bcls --minimum-trimmed-read-length 0 --mask-short-adapter-reads 0
Generate Undetermined fastq¶
# if you haven't done hpcf_interactive, run the next command
hpcf_interactive -q standard -R "rusage[mem=32000]"
module load bcl2fastq
wget https://raw.githubusercontent.com/YichaoOU/HemTools/master/docs/gallery/minimal_SampleSheet.csv
bcl2fastq --no-lane-splitting -o raw_fastq --sample-sheet minimal_SampleSheet.csv
Generate R1 and R2 reads with I1 and I2 in the read name¶
Header example:
@M04990:107:000000000-JYJGM:1:1101:20373:1000 1:N:0:NCTCCGAC+NTANNNCNNNNNNNNTTC
By default, `bcl2fastq indeed does put index reads in the read name. However, it is only when you have some samples specified in the SampleSheet.csv. To get Undertermined R1, R2, I1, I2 with read name containing index, you need to have some dummy SampleSheet.csv.
# if read length is 301bp, uditas, I8, I18
bcl2fastq --no-lane-splitting -o fastq_files --sample-sheet /research/dept/hem/common/sequencing/210109_M04990_0003_000000000-J87CP/Data/Intensities/BaseCalls/SampleSheet.csv --create-fastq-for-index-reads
# uditas, 151 read, I8, I18
bcl2fastq --no-lane-splitting -o fastq_files --sample-sheet /home/yli11/HemTools/share/misc/SampleSheet_I8_I18.csv --create-fastq-for-index-reads
# if read length is 151bp
bcl2fastq --no-lane-splitting -o fastq_files --sample-sheet /home/yli11/HemTools/share/misc/SampleSheet.csv --create-fastq-for-index-reads
# for starr-seq
bcl2fastq --no-lane-splitting -o starr_seq_fastq --sample-sheet /home/yli11/HemTools/share/misc/starr_seq_SampleSheet.csv --create-fastq-for-index-reads
# for starr-seq demultiplexing
starr_seq_demultiplex.py ATTACTCG TATAGCCT ATAGAGGC 1
# DNA barcode R1, R2, RNA barcode, mismatch cutoff
# for g34 iSeq
cp /home/yli11/dirs/hem_seq/20220505_FS10001356_32_BPN80008-1429/SampleSheet.csv .
bcl2fastq --no-lane-splitting -o fastq_files
Sequence data is paired-end with 8bp i7 and i5, but my actual data is just single-end read with just index 1 6bp.¶
In this case, you don’t need to do anything different for bcl2fastq command. You just need to only put i7 6bp index in the SampleSheet.csv file. After bcl2fastq, you will have R1 and R2 reads, but you only need R1 reads.
Comments¶
code @ github.