Extract inward/outward oriented pairs from BAM file¶
usage: extract_reads.py [-h] [-o OUTPUT] [-f BAM_FILE] (--outward | --inward)
optional arguments:
-h, --help show this help message and exit
-o OUTPUT, --output OUTPUT
output table name (default:
extract_reads_2020-03-11.csv)
-f BAM_FILE, --bam_file BAM_FILE
input bam file (default: None)
--outward get outward reads (default: False)
--inward get inward reads (default: False)
Summary¶
This program can extract inward or outward reads from a given BAM file. This problem will only consider properly paired reads and not supplementary alignments. Results are matched to the numbers from samtools stats.
Inward read, or innie, is defined as the read that’s mapped to the forward strand comes before the read mapped to the reverse strand when their 5’-end coordinates are compared.
Outward read, or outie, is defined as the read that’s mapped to the forward strand comes after the read mapped to the reverse strand when their 5’-end coordinates are compared.
Definition can be found here: https://github.com/samtools/htsjdk/blob/master/src/main/java/htsjdk/samtools/SamPairUtil.java
The definitions from samtools seem to be a little bit different:
inward oriented pairs - number of paired reads with flag 0x40 (64) set and flag 0x10 (16) not set or with flag 0x80 (128) set and flag 0x10 (16) set.
outward oriented pairs - number of paired reads with flag 0x40 (64) set and flag 0x10 (16) set or with flag 0x80 (128) set and flag 0x10 (16) not set.
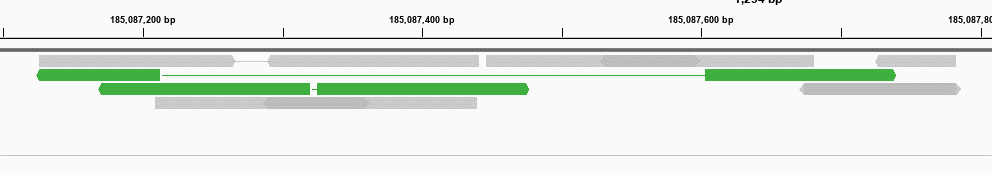
Examples of outward oriented pairs are highlighted in green.
Input¶
A bam file with index in the same dir.
Usage¶
hpcf_interactive
module load python/2.7.13
extract_reads.py -f input.bam --outward -o output.csv
To get inward reads, you can use --inward option.
Output¶
Output file contains 3 columns: read name, read 1 sequences, read 2 sequences.
FAQ¶
Numbers do not match?¶
Remember that in this program, reads are filtered. To get the number match, you can filter your bam file like below:
samtools view -b -F 2048 -f 2 input.bam > test.bam
samtools index test.bam
samtools stats test.bam | head -37
extract_reads.py -f test.bam --outward
Comments¶
code @ github.