Plot Venn diagram given two bed files¶
Please use intervene [here](https://intervene.readthedocs.io/en/latest/examples.html)
hpcf_interactive
source activate /home/yli11/.conda/envs/py2
module load R/3.5.1
intervene upset -i *unmatched.realign.tsv
Result is stored in a folder called Intervene_results
OLD notes¶
usage: bed2venn.py [-h] -b1 B1 -b2 B2 -l1 L1 -l2 L2 [-e1 E1] [-e2 E2]
[--fontsize FONTSIZE] [-o OUTPUT]
optional arguments:
-h, --help show this help message and exit
-b1 B1 bed file 1 (default: None)
-b2 B2 bed file 2 (default: None)
-l1 L1 label 1 (default: None)
-l2 L2 label 2 (default: None)
-e1 E1 extend b1 bed file by e1 bp (default: None)
-e2 E2 extend b2 bed file by e2 bp (default: None)
--fontsize FONTSIZE label 2 (default: 10)
-o OUTPUT, --output OUTPUT
output file name (default: yli11_2021-04-26)
Summary¶
This program will calculate the amount of overlaps given two bed files.
Note that, it is very likely that the number of A overlapping with B is different than the number of B overlapping with A.
For example, let’s say A is an entire chromosome, B is some scattered regions in this chromosome. Then, the number of A overlapping with B is 1 and the number of B overlapping with A is |B|.
Input¶
Two bed files. Any bed formats that are supported by bedtools are acceptable here. e.g., narrowpeak files.
Output¶
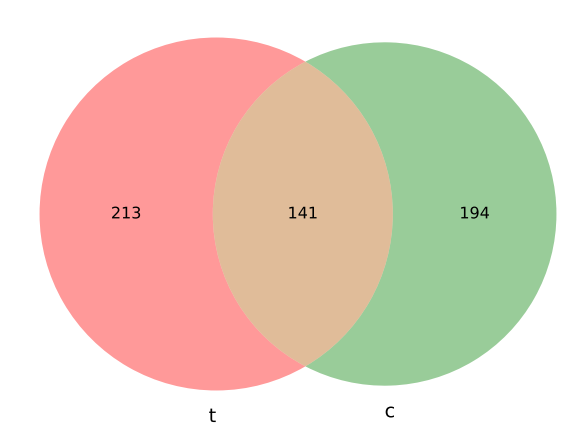
Usage¶
hpcf_interactive
module load python/2.7.13
bed2venn.py -b1 bed1 -b2 bed2 -l1 treatment -l2 control
To extend Xbp left and right on -b1 bed file or -b2 bed file, please use -e1 or -e2
hpcf_interactive
module load python/2.7.13
bed2venn.py -b1 bed1 -b2 bed2 -l1 treatment -l2 control -e1 5000