Heatmap Basic¶
usage: plot_heatmap.py [-h] [-j JID] [--remove_zero] [--index] -f INPUT
[--index_using INDEX_USING] [-s SEP] [--xlabel XLABEL]
[--ylabel YLABEL] [--title TITLE]
[--figure_type FIGURE_TYPE] [--remove_cols REMOVE_COLS]
[-o OUTPUT] [--header] [--no_col_names]
[--no_row_names] [-W WIDTH] [-H HEIGHT]
[--just_default] [--log2_transform] [--just_plot]
[--top_N_by_row_mean TOP_N_BY_ROW_MEAN]
[--row_mean_cutoff ROW_MEAN_CUTOFF]
[--row_mean_percent_col_cutoff ROW_MEAN_PERCENT_COL_CUTOFF [ROW_MEAN_PERCENT_COL_CUTOFF ...]]
plot heatmap given dataframe.
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
plot_heatmap_yli11_2020-04-07)
--remove_zero remove all rows or cols that are zero (default: False)
--index index is false (default: False)
-f INPUT, --input INPUT
data table input (default: None)
--index_using INDEX_USING
Sometimes we want to show a different label for the
indices, then use this option. For example, gene id is
unique, however, gene name can be different, it also
has upper or lower case problem, in such case, we want
to use gene id for data processing and use gene name
for visualization. (default: )
-s SEP, --sep SEP separator (default: )
--xlabel XLABEL
--ylabel YLABEL
--title TITLE
--figure_type FIGURE_TYPE
pdf,png,jpeg (default: png)
--remove_cols REMOVE_COLS
-o OUTPUT, --output OUTPUT
output table name (default: yli11_2020-04-07)
--header input table has header (default: False)
--no_col_names Don't show column names in the heatmap (default:
False)
--no_row_names Don't show row names in the heatmap (default: False)
-W WIDTH, --width WIDTH
Figure width, by default, w=N_row/4, if given, will
replace the default value (default: )
-H HEIGHT, --height HEIGHT
Figure height, by default, w=N_col/4, if given, will
replace the default value (default: )
--just_default just plot using default seaborn parameters (default:
False)
--log2_transform input values will be log2 transformed (default: False)
--just_plot with this option, no filters will be applied. This
program will just plot a heatmap based on the input
dataframe (default: False)
Applying row filters to your dataframe, heatmaps will be generated for each of the following filters.:
--top_N_by_row_mean TOP_N_BY_ROW_MEAN
plot the top N rows (default: 50)
--row_mean_cutoff ROW_MEAN_CUTOFF
plot the rows with a minimal mean value (default: 5)
--row_mean_percent_col_cutoff ROW_MEAN_PERCENT_COL_CUTOFF [ROW_MEAN_PERCENT_COL_CUTOFF ...]
row mean cutoff, col fraction cutoff, space separated
(default: [5, 0.1])
Summary¶
This is for basic heatmap, without additional row/col highlights.
Given a 2D data, plot heatmaps for several filters and several parameters. See above description for row and col filters. See ClusterMap for clustermap parameters, such as linkage method (e.g., average) and row/col normalization. For all the figures, take the one that is most useful to you. If you just want to plot the input data without any filters, use --just_plot.
distance metric used: euclidean (default), cosine, correlation.
linkage method used: average (default), ward.
Not all available choices are used. Just use some typical ones. Otherwise there will be so many figures to look at (because it is combinatorial!). This combination, together with row/col normalization, will generate 16 figures for the same input dataframe. You can turn of this option using --just_default.
Example¶
Input¶
Data table, tsv (default) or csv (-s ,). If data table contains both row names and column names, use --index --header
Usage¶
hpcf_interactive -q standard -R "rusage[mem=20000]"
module load conda3
source activate /home/yli11/.conda/envs/py2/
Example usage I: plot the raw data
My input is data.csv, so I need to set the separator to be ,. For raw data, since it is very big 5000*5000, then it is better to turn of xtick labels, which is --no_col_names --no_row_names. I want to do log transform because my input is gene counts, --log2_transform. Since I just want to get a sense of the data, I don’t need to see very pixel, so I set image size as -W 20 -H 20.
Note
If N rows * N cols > 1M, we will just take a random sample of row or col for heatmap with N=1000. The reason we can’t do larger size is because of the recursion limit for clustering dendrogram. One way to solve it is using other method to sort rows and columns, get a new dataframe, and just plot this dataframe without row/col clustering.
plot_heatmap.py -f data.csv -o Banana --log2_transform --just_plot -W 20 -H 20 --header --index --no_col_names --no_row_names -s ,
Example usage II: plot filtered data
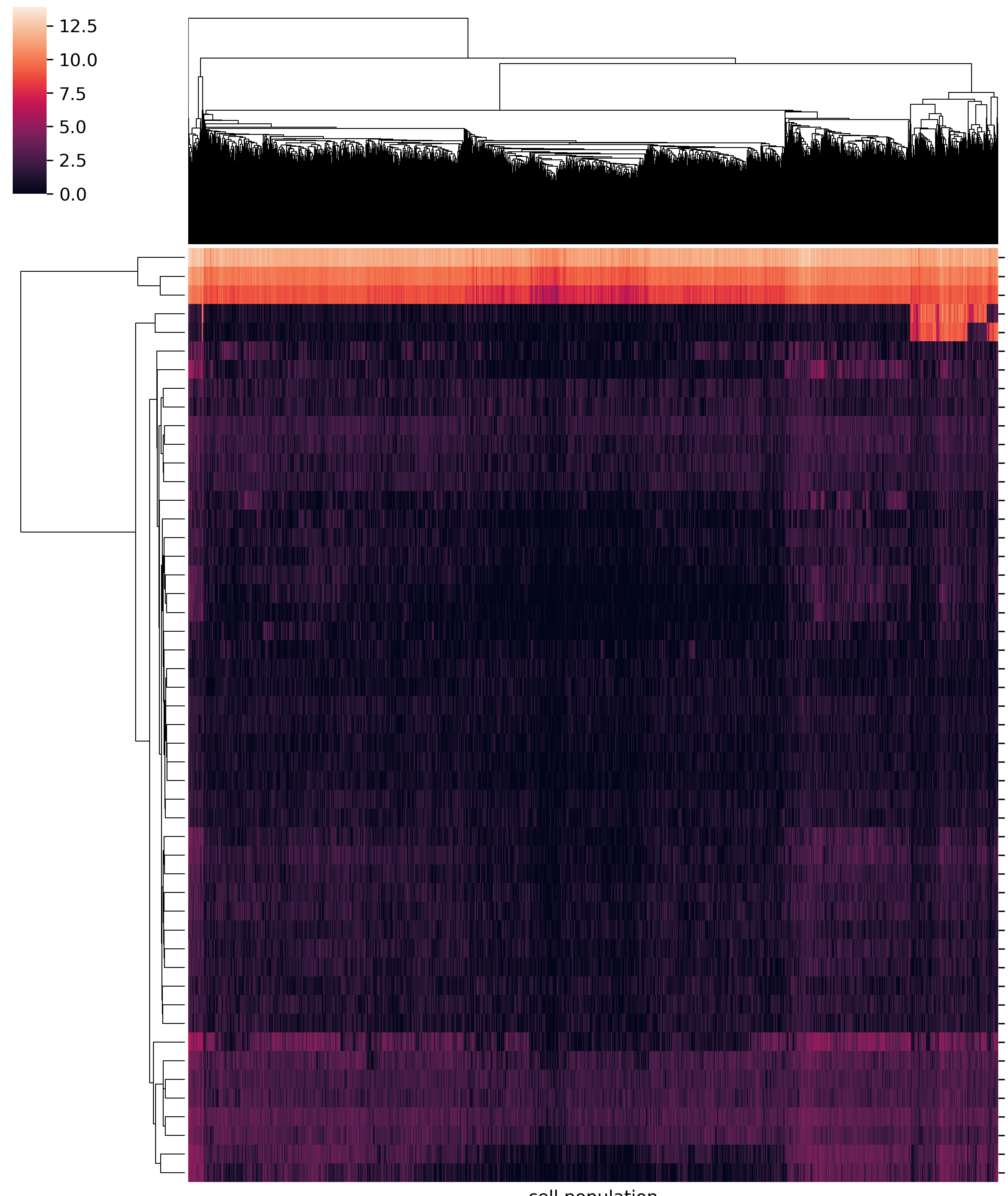
Here, I’m plotting single-cell RNA-seq data. The data table returned by cellranger uses genes as rows and cells as columns. I’m not interested in knowing the barcode, so I turned of col names by --no_col_names. I also set -W 10, since we have so many cells, by default the width could be very large. My data table contains both row names and column names, so use --index --header.
plot_heatmap.py -f data.csv -o Banana --log2_transform -W 10 -s , --index_using genes.tsv --no_col_names --index --header --xlabel "cell population" --ylabel "genes" --title Banana
Tip
If you want pdf files, use --file_type pdf. Generating pdf files are slow for large matrices! At least 10 min for 1K*1K data.
Output¶
All figures will be generated in the job ID folder. Look at *default* figures. If they don’t look interesting, then go to “others” folder, there will be 16 figures for the same df using different parameter combinations.
Comments¶
code @ github.