MicroC data analysis pipeline¶
usage: MicroC.py [-h] [-j JID] [-f FASTQ_TSV] [-q QUEUE] [-m MEMORY]
[--FLASH_min_overlap FLASH_MIN_OVERLAP]
[--FLASH_max_overlap FLASH_MAX_OVERLAP] [--src SRC]
[-g GENOME] [--bowtie2_index BOWTIE2_INDEX]
[--genome_fasta GENOME_FASTA] [-s CHROM_SIZE] [--interactive]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
MicroC_yli11_2022-02-08)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
4 columns: R1, R2, label, bait.bed (default: None)
-q QUEUE, --queue QUEUE
submit queue (default: standard)
-m MEMORY, --memory MEMORY
request memory (default: 30000)
--FLASH_min_overlap FLASH_MIN_OVERLAP
FLASH_min_overlap (default: 10)
--FLASH_max_overlap FLASH_MAX_OVERLAP
FLASH_max_overlap (default: 100)
--src SRC src of MicroC program (default: /home/yli11/Programs
/Micro-Capture-C)
--interactive run pipeline interatively (default: False)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
--bowtie2_index BOWTIE2_INDEX
fasta index (default:
/home/yli11/Data/Human/hg19/index/bowtie2_index/hg19)
--genome_fasta GENOME_FASTA
fasta (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
-s CHROM_SIZE, --chrom_size CHROM_SIZE
chrome size (default: /home/yli11/Data/Human/hg19/anno
tations/hg19.chrom.sizes)
MicroC worflow¶
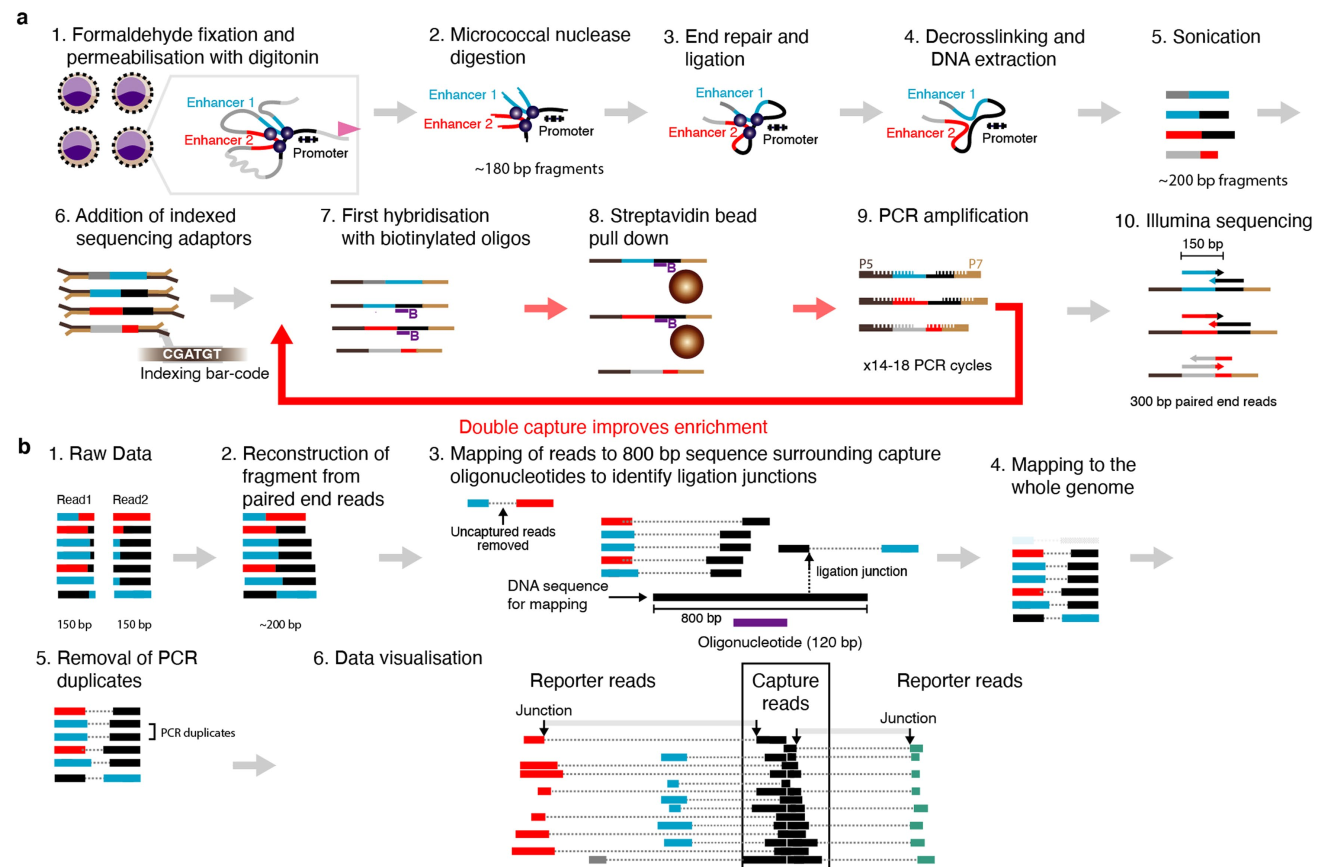
Summary¶
Pipeline adopted and customized from https://github.com/jojdavies/Micro-Capture-C
Warning
Only work for hg19/mm9 right now, by 2/16/2022.
Input¶
Example: Sequencing_runs/chenggrp_253074_HiC-1_MCC/analysis
Target bait bed file
Need 4 columns, chr, start, end, name
Warning
The bait name in the last column should not contain “_”. This char is used in the MCC code as a key separator.
==> HBG1_bait.bed <==
chr11 5272143 5272263 gene1
chr11 5270668 5270788 gene2
==> HBG2_bait.bed <==
chr11 5277094 5277214 ABC
chr11 5275592 5275712 DEF
Note
You can have either (1) multiple loci baits in one bed file or (2) only one locus baits one bed file. If (1), then the merged results (a merged bw file generated by the pipeline) will make no sense.
fastq.tsv
Need 4 columns, R1, R2, label, bait bed file name
Use --guess_input to automatically generate the first 3 columns. Here I manually edited the 3rd column.
8_S15_L002_R1_001.fastq.gz 8_S15_L002_R2_001.fastq.gz H2_15_HBG2 HBG2_bait.bed
7_S14_L002_R1_001.fastq.gz 7_S14_L002_R2_001.fastq.gz H2_15_HBG1 HBG1_bait.bed
6_S13_L002_R1_001.fastq.gz 6_S13_L002_R2_001.fastq.gz H2_10_HBG2 HBG2_bait.bed
5_S12_L002_R1_001.fastq.gz 5_S12_L002_R2_001.fastq.gz H2_10_HBG1 HBG1_bait.bed
4_S11_L002_R1_001.fastq.gz 4_S11_L002_R2_001.fastq.gz H1_15_HBG2 HBG2_bait.bed
3_S10_L002_R1_001.fastq.gz 3_S10_L002_R2_001.fastq.gz H1_15_HBG1 HBG1_bait.bed
2_S9_L002_R1_001.fastq.gz 2_S9_L002_R2_001.fastq.gz H1_10_HBG2 HBG2_bait.bed
1_S8_L002_R1_001.fastq.gz 1_S8_L002_R2_001.fastq.gz H1_10_HBG1 HBG1_bait.bed
Note
The 3rd column should be unique, each run, the output will put into the 3rd. So if they are not unique, the next run will overwrite previous results. If for the same fastq file, you have multiple loci, then you have to write down that fastq files into multiple lines, each line for each locus.
Usage¶
hpcf_interactive
module load python/2.7.13
run_lsf.py --guess_input # to generate fastq.tsv
# Please manually add the 4th column to fastq.tsv before running this
MicroC.py -f fastq.tsv -g hg19
QC¶
These reports are attached in the email notification.
multiqc_report.html¶
General statistics, in this section, we may look at Length, M Seqs, and %Combined; average read length, total read count, and percentage of combined reads.
FastQC, in this section, we may look at Sequence Quality Histograms.
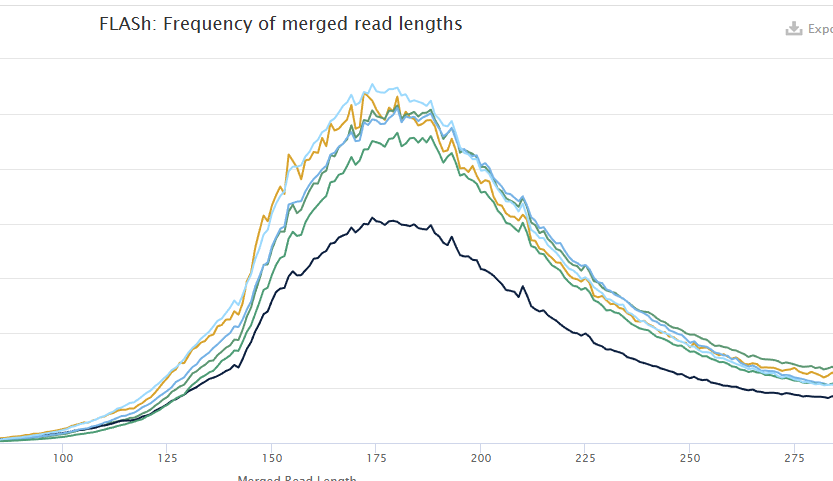
FLASH, we can look at the fragment length distribution. Should be a broad peak around 170-200bp
MicroC_summary.html¶
In the summary table, we can look at capture efficiency (ranging from 3% to 20%), read duplication rate (usually very high, ~95%) and number of unique interactions. If number of unique interactions less than 10k, might consider sequencing more. >100k is consdered to be deep.
Some numbers¶
Third, we generated extremely deep data from individual viewpoints (up to 500,000 unique contacts (mean, 140,000) per 120-bp viewpoint) (Supplementary Table 1). This equates to more than 1,000-fold the depth of data obtained with ‘all versus all’ approaches such as Hi-C and Micro-C (more than 3 trillion ligation junctions would be required for this depth of coverage genome-wide).
Output¶
In the jobID folder, result is organized for each sample (*_read_split folder). In each folder, you can find bw files:
L_H1_10_HBG1_ALL_FP.bw
L_H1_10_HBG1_UP_FP.bw
R_H1_10_HBG1_DO_FP_plusone.bw
L_H1_10_HBG1_ALL_FP_plusone.bw
L_H1_10_HBG1_UP_FP_plusone.bw
R_H1_10_HBG1.read_pileup.bw
L_H1_10_HBG1_de_norm_rep.bw
R_H1_10_HBG1_ALL_FP.bw
R_H1_10_HBG1_UP_FP.bw
L_H1_10_HBG1_DO_FP.bw
R_H1_10_HBG1_ALL_FP_plusone.bw
R_H1_10_HBG1_UP_FP_plusone.bw
L_H1_10_HBG1_DO_FP_plusone.bw
R_H1_10_HBG1_de_norm_rep.bw
L_H1_10_HBG1.read_pileup.bw
R_H1_10_HBG1_DO_FP.bw
Known Problem¶
FASTQC has a memory upper limit that we can’t set. Need to manually kill the process to let the pipeline continue
Custom Genome¶
custom genome is supported, you have to use -g custom then give the absolute path to --bowtie2_index --genome_fasta --chrom_size
Ref¶
https://www.nature.com/articles/s41586-021-03639-4