ATAC-seq¶
Summary¶
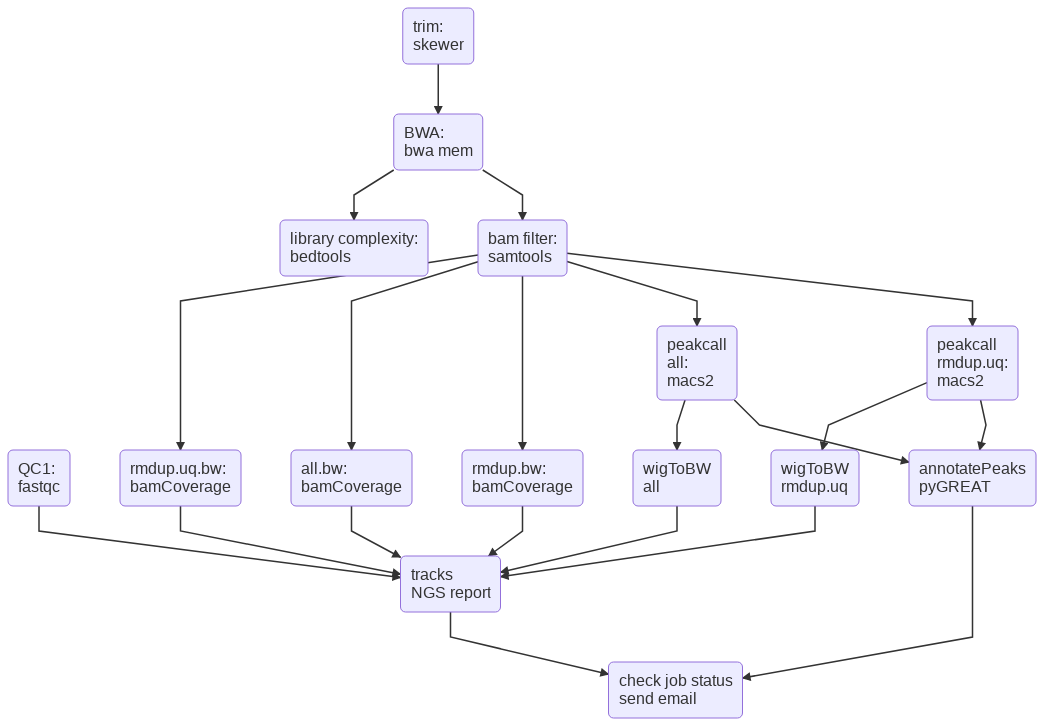
The ATAC-seq pipeline first trims the raw reads to remove Tn5 adaptor sequence using skewer. Then reads are mapped to the genome (-g) using BWA mem. Raw mapped reads are labeled as .markdup.bam. De-duplicated reads are labeled as .rmdup.bam. De-duplicated and uniquely mapped reads are labeled as .rmdup.uq.bam. Duplicated and multi-mapped reads were removed using samtools (v0.17). ATAC-seq peaks were called using MACS2 (v2.1.1) with the following parameters “macs2 callpeak –nomodel –shift -100 –extsize 200”. BigWiggle files were generated using DeepTools bamCoverage (v3.2.0) with “–centerReads”.
Code for this pipeline is provided in “https://github.com/YichaoOU/HemTools/blob/master/subcmd/”. See atac_seq.py and utils.py.
Parameters¶
usage: HemTools atac_seq [-h] [-j JID] [--short] [--debug]
(-f INPUT | --guess_input) [-i INDEX_FILE]
[-g GENOME] [-b BLACKLIST] [-s CHROM_SIZE]
[-e EFFECTIVEGENOMESIZE]
Named Arguments¶
- -j, --jid
enter a job ID, which is used to make a new directory. Every output will be moved into this folder.
Default:
'{{subcmd}}_docs_2026-04-30'- --short
Force to use the short queue. (only if R1+R2 fastq.gz size <=250M)
Default:
False- --debug
Not for end-user.
Default:
False- -f, --input
tab delimited 3 columns (tsv file): Read 1 fastq, Read 2 fastq, sample ID
- --guess_input
Let the program generate the input files for you.
Default:
False
Genome Info¶
- -i, --index_file
BWA index file
Default:
'/home/docs/checkouts/readthedocs.org/user_builds/hemtools/checkouts/latest/subcmd/../hg19/bwa_16a_index/hg19.fa'- -g, --genome
genome version: hg19, hg38, mm10, mm9.
Default:
'hg19'- -b, --Blacklist
Blacklist file
Default:
'/home/docs/checkouts/readthedocs.org/user_builds/hemtools/checkouts/latest/subcmd/../hg19/Hg19_Blacklist.bed'- -s, --chrom_size
chrome size
Default:
'/home/docs/checkouts/readthedocs.org/user_builds/hemtools/checkouts/latest/subcmd/../hg19/hg19.chrom.sizes'- -e, --effectiveGenomeSize
effectiveGenomeSize for bamCoverage
Default:
'2451960000'
Flowchart¶
Usage¶
Go to your data directory and type the following.
Step 0: Load python version 2.7.13.
$ module load python/2.7.13
Step 1: Prepare input files, generate fastq.tsv.
$ HemTools atac_seq --guess_input
Input fastq files preparation complete! ALL GOOD!
Please check if you like the computer-generated labels in : fastq.tsv
Note
If you are preparing fastq.tsv yourself, please make sure no space anywhere in the file. Note that the seperator is tab. Spaces in file name will cause errors.
Step 2: Check the computer-generated input list (manually), make sure they are correct.
$ less fastq.tsv
Note
a random string will be added to the generated files (e.g., fastq.94c049cbff1f.tsv) if they exist before running step 1.
Step 3: Submit your job.
$ HemTools atac_seq -f fastq.tsv
Sample input format¶
fastq.tsv
This is a tab-seperated-value format file. The 3 columns are: Read 1, Read 2, sample ID.

Quality Control¶
The quality metrics are provided in the html report. For ChIP-seq data, we also provide strand cross-correlation metrics (i.e., those attached pdf files).
Metrics |
Threshold |
NRF |
>0.9 |
PBC1 |
>0.9 |
PBC2 |
>3 |
Num peaks |
>100k |
https://www.encodeproject.org/atac-seq/
https://www.encodeproject.org/chip-seq/transcription_factor/ https://www.encodeproject.org/chip-seq/histone/
https://github.com/crazyhottommy/ChIP-seq-analysis/blob/master/part0_quality_control.md
Report bug¶
Once the job is finished, you will be notified by email with some attachments. If no attachment can be found, it might be caused by an error. In such case, please go to the result directory (where the log_files folder is located) and type:
$ HemTools report_bug
Use different genome index¶
$ HemTools atac_seq -f fastq.tsv -i YOUR_GENOME_INDEX
Example of using different genome index¶
Ruopeng masked index
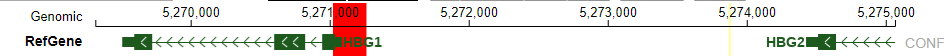
HemTools atac_seq -f fastq.tsv -i /home/yli11/Data/Human/hg19/index/masked_genome/ruopeng_hbg1_promoter/ruopeng_hbg1_promoter.mask.fa
Li masked index

HemTools atac_seq -f fastq.tsv -i /home/yli11/Data/Human/hg19/index/masked_genome/li_hgb1_promoter/li_hgb1_promoter.mask.fa
Comments¶
code @ github.