PacBio iso-seq data analysis¶
Summary¶
Pipeline adopted from ENCODE Long Read RNA-Seq Analysis Protocol for Human Samples (v3.0): https://www.encodeproject.org/documents/81af563b-5134-4f78-9bc4-41cb42cc6a48/@@download/attachment/ENCODE%20Long%20Read%20RNA-Seq%20Analysis%20Pipeline%20v3%20%28Human%29.pdf
Only work for hg38 right now, by 6/6/2022.
Current pipeline doesn’t filter/merge based on replicates.
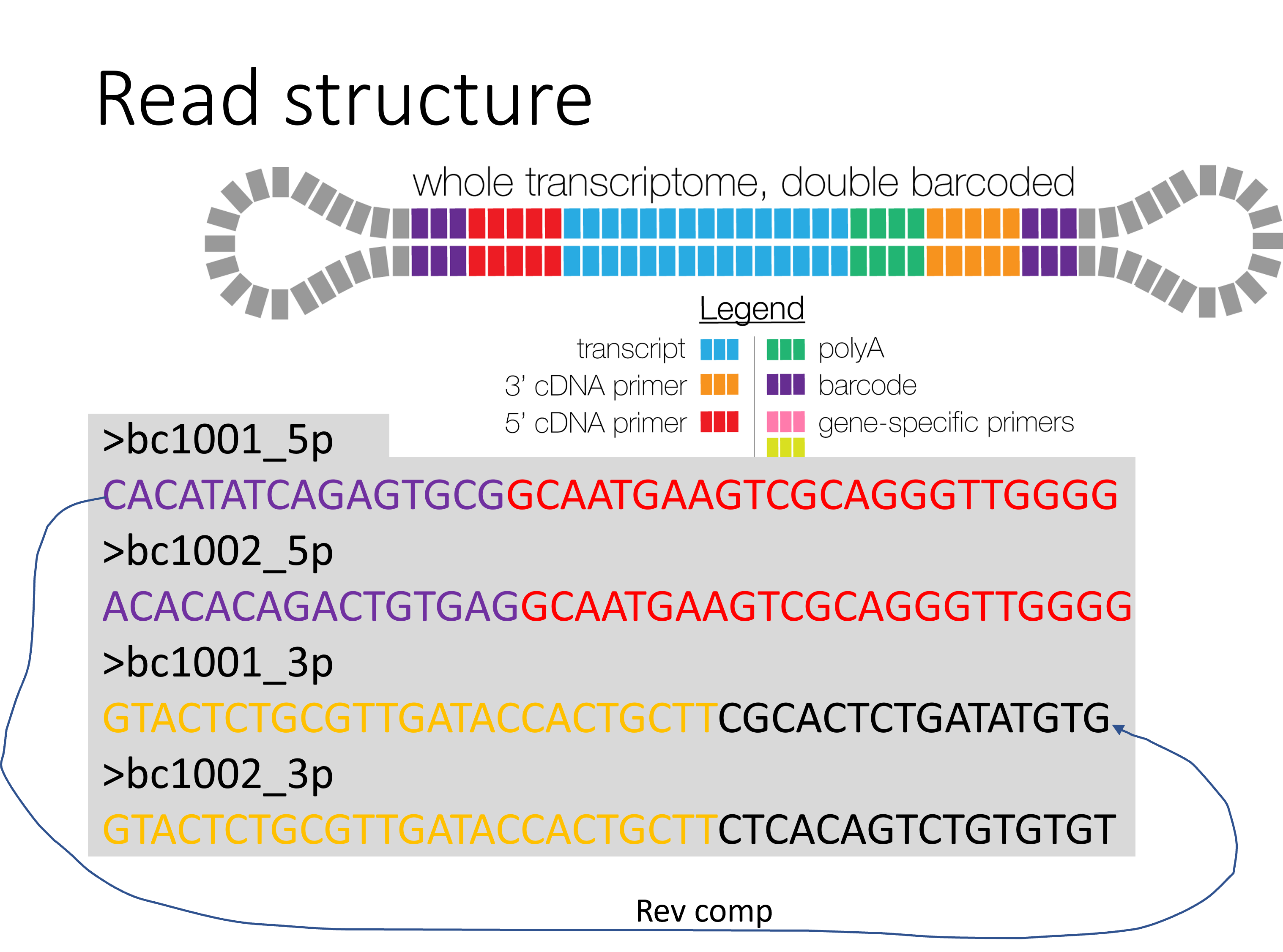
Iso-seq read structure¶
Here at St. Jude we use a double barcoded primers, each sample has the unique barcode, 5p and 3p primers have the identical but reverse completed barcodes.
Input¶
input.list¶
A 2-column tsv file, the first column is PacBio hifi reads, the last column is sample ID, should be unique.
control.bam control
KO.bam KO
primer.fa¶
>primer_5p
GCAATGAAGTCGCAGGGTTGGGG
>primer_3p
GTACTCTGCGTTGATACCACTGCTT
Usage¶
hpcf_interactive
module load python/2.7.13
run_lsf.py -f input.list --primer primer.fa -p iso_seq
Output¶
Results can be found in the $jid folder.
1. raw read annotation file¶
*final_talon_read_annot.tsv
2. transcript/gene abundance file (main results)¶
*final_talon_abundance.tsv
3. custom gene annotation file¶
*talon.gtf
Reference¶
Notes¶
To resume failed runs, submit the job with the same jid -j and also use --override_jid