CaptureC data analysis pipeline¶
usage: captureC.py [-h] [-j JID] (--guess_input | -f FASTQ_TSV) [-t TARGET]
[-l EXCLUSION_LENGTH] [-g GENOME] [-i INDEX_FILE]
[-s CHROM_SIZE] [-fa GENOME_FA] [-e DIGESTED_ENZYME]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
captureC_yli11_2021-05-04)
--guess_input Let the program generate the input files for you.
(default: False)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
3 columns (default: None)
-t TARGET, --target TARGET
9 columns (default: None)
-l EXCLUSION_LENGTH, --exclusion_length EXCLUSION_LENGTH
exclusion_length (default: 1000)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
-i INDEX_FILE, --index_file INDEX_FILE
BWA index file (default: /home/yli11/Data/Human/hg19/i
ndex/bowtie_1.2.2_CapC_index)
-s CHROM_SIZE, --chrom_size CHROM_SIZE
chrome size (default: /home/yli11/Data/Human/hg19/anno
tations/hg19.chrom.sizes)
-fa GENOME_FA, --genome_fa GENOME_FA
Blacklist file (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
-e DIGESTED_ENZYME, --digested_enzyme DIGESTED_ENZYME
digested_fragments hg19_MboI (default: MboI)
Summary¶
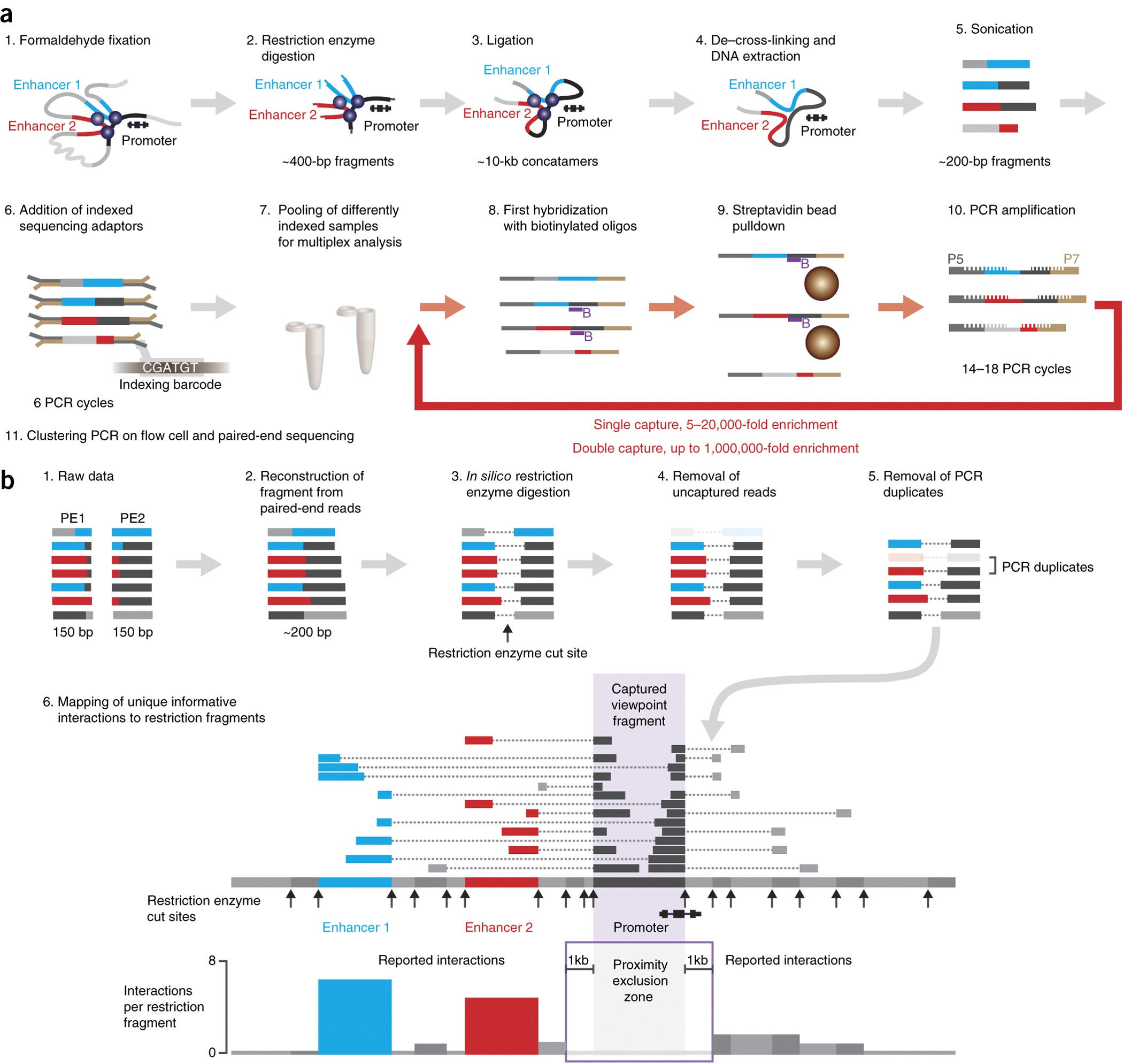
Pipeline adopted from https://github.com/Hughes-Genome-Group/captureC
Only work for hg19 right now, by 5/4/2021.
This pipeline produce similar results to hicpro pipeline. However, when dealing with HBG1/HBG2 data, this pipeline appears to be better.
Input¶
fastq.tsv
Use --guess_input to automatically generate this.
Banana_R1.fastq.gz Banana_R2.fastq.gz Banana_lovers
Orange_R1.fastq.gz Orange_R2.fastq.gz Orange_lovers
Target bait bed file
at least 3 columns
chr1 123 456
chr1 454 654
Usage¶
hpcf_interactive
module load python/2.7.13
captureC.py --guess_input # to generate fastq.tsv
captureC.py -f fastq.tsv -t target.bed
Default digested enzyme is MboI, which has the same cutting site as dpnII. If you used NlaIII, then please use:
captureC.py -f fastq.tsv -t target.bed -e NlaIII -g hg19
Others¶
To make a list of dpnII cut sites:
Differential analysis or data normalization¶
The pipeline output DESEQ2.input.tsv in jid folder.
You can use it as the input for DESEQ2 pipeline. run_DESEQ2.py
Reference¶
Notes¶
Plan to update the current pipeline to: https://github.com/Hughes-Genome-Group/CCseqBasicS
ref: https://www.nature.com/articles/s41467-019-13404-x
Input OLD¶
fastq.tsv
Use --guess_input to automatically generate this.
Banana_R1.fastq.gz Banana_R2.fastq.gz Banana_lovers
Orange_R1.fastq.gz Orange_R2.fastq.gz Orange_lovers
Target bait file (MUST end with
.txt)
Need absolute path to this file
Columns are: Name, chr, target_start, target_end, chr, exclusion_start, exclusion_end, 1, A.
The last two columns are almost always 1 A, which means that I don’t have a SNP defined.
Make sure there’s no empty row in this file.
HS3 11 5305797 5306271 11 5304797 5307271 1 A