MNase-based HiCHIP data analysis¶
usage: hichip.py [-h] [-j JID] -f FASTQ_TSV -p PEAK_BED [-IntType INTTYPE]
[-BINSIZE BINSIZE] [-mapq_cutoff MAPQ_CUTOFF]
[-LowDistThr LOWDISTTHR] [-UppDistThr UPPDISTTHR]
[-UseP2PBackgrnd USEP2PBACKGRND] [-BiasType BIASTYPE]
[-MergeInt MERGEINT] [-QVALUE QVALUE] [-g GENOME]
[--bwa_index BWA_INDEX] [-s CHROM_SIZE]
RGT_HINT atac-seq footprint with bias correction
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
hichip_yli11_2022-08-01)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
3-col tsv, R1, R2, sample_ID (default: None)
-p PEAK_BED, --peak_bed PEAK_BED
narrowpeak file (default: None)
-IntType INTTYPE #Interaction type - 1: peak to peak (CTCF) 2: peak to
non peak 3: peak to all (default, h3k27ac) 4: all to
all 5: everything from 1 to 4. (default: 3)
-BINSIZE BINSIZE # Size of the bins, in bases, for detecting the
interactions. (default: 2500)
-mapq_cutoff MAPQ_CUTOFF
mapq_cutoff (default: 0)
-LowDistThr LOWDISTTHR
Lower distance threshold of interaction between two
segments (default: 10000)
-UppDistThr UPPDISTTHR
Upper distance threshold of interaction between two
segments (default: 2000000)
-UseP2PBackgrnd USEP2PBACKGRND
# Applicable only for peak to all output interactions
- values: 0 / 1 # if 1, uses only peak to peak loops
for background modeling - corresponds to FitHiChIP(S)
# if 0, uses both peak to peak and peak to nonpeak
loops for background modeling - corresponds to
FitHiChIP(L) (default: 1)
-BiasType BIASTYPE # parameter signifying the type of bias vector -
values: 1 / 2 # 1: coverage bias regression 2: ICE
bias regression (default: 1)
-MergeInt MERGEINT # following parameter, if 1, means that merge
filtering (corresponding to either FitHiChIP(L+M) or
FitHiChIP(S+M)) # depending on the background model,
would be employed. Otherwise (if 0), no merge
filtering is employed. (default: 1)
-QVALUE QVALUE FDR (q-value) threshold for loop significance
(default: 0.05)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, hg38, mm9, mm10. By default,
specifying a genome version will automatically update
index file, black list, chrom size and
effectiveGenomeSize, unless a user explicitly sets
those options. (default: hg19)
--bwa_index BWA_INDEX
bwa index file (default: /home/yli11/Data/Human/hg19/i
ndex/bwa_16a_index/hg19.fa)
-s CHROM_SIZE, --chrom_size CHROM_SIZE
chrome size (default: /home/yli11/Data/Human/hg19/anno
tations/hg19.chrom.sizes)
Summary¶
Pipeline is adopted from: https://hichip.readthedocs.io/en/latest/index.html
Input¶
Requires paired-end fastq files and a peak (narrowPeak format) file. If we are doing a CTCF HiChIP, then we should have done a CTCF ChIP-seq in order to have the CTCF narrowPeak file.
Usage¶
Go to your fastq files folder and do the following:
hpcf_interactive
module load python/2.7.13
run_lsf.py --guess_input
hichip.py -f fastq.tsv -p CTCF.bed -g hg19 -IntType 1
hichip.py -f fastq.tsv -p H3k27ac.bed -g hg19 -IntType 3
1 means peak-to-peak interaction 3 means peak-to-all interaction (see help message for other types 1-5)
Output¶
1. Library QC¶
See hichip_qc_summary.html. QC standards are:
Metric |
Shallow Seq (20M) |
Deep Seq (100-200M) |
|---|---|---|
No-Dup Read Pairs |
>75% |
>50% |
No-dup cis read pairs ≥ 1kb |
>20% |
>20% |
Total reads in 1000 bp around center of peaks |
>2% |
>2% |
NOTE: The FitHiCHIP author said if ``No-dup cis read pairs ≥ 10kb `` is around 10M, you should increase bin size (i.e., lower resolution) to 20kb in order to see more significant loops. I found if the value is near 50M, then 2.5kb or 5kb bin is OK; I’m aiming for 10k to 20k total interactions.
I also found this GATA1 HiCHIP data in mouse https://www.pnas.org/doi/full/10.1073/pnas.2008672117 where they used juicer tools and identified 40K-80K interactions (I guess these are ALL to ALL interactions).
To just redo fithichip calling, modify the config file (hichip.config.txt) in the jobID folder and run the following:
Remember to change OutDir, otherwise the default output folder fithichip_results will be overwritten.
bsub -q priority -P Genomics -R 'rusage[mem=30000]' -J fit /home/yli11/Programs/FitHiChIP/FitHiChIP_HiCPro.sh -C hichip.config.txt
2. Called interactions¶
*fithic.merged.counts.bedpe and *fithic.merged.pvalue.bedpe
FitHiChIP detailed results are provided in fihichip_results. It also provides a result summary html file.
For fitHiCHIP called interactions, 10k to 20k are already good numbers (Q value 0.05) because this tool is quite stringent.
The above files contain merged loops. We found the unmerged interactions seem to be better (more sensitive). You can find them in folders like fithichip_results/FitHiChIP_Peak2ALL_{binSize}_L10000_U2000000/P2PBckgr_0/Coverage_Bias/FitHiC_BiasCorr/*.interactions_FitHiC_{Q-value}.bed
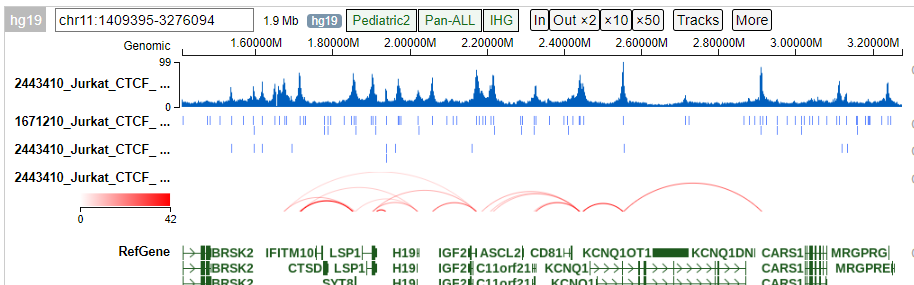
3. Tracks visualization¶
In the upload folder, users can find:
mapped.PT.bw: read pairs bw files
macs2_peaks.narrowPeak: de-novo called peaks from HiCHIP data
fithic.merged.counts.bedpe: FitHiChIP called interactions
user-provided peak file
Users can run create_tracks.py --current_dir -g $genome to upload these files to protein paint.
Redo interaction calling¶
Modify parameters in hichip.config.txt and rerun Fit-hichip
First, load dependencies:
module load conda3/202011
source activate /home/yli11/.conda/envs/HiChIP
module load bwa/0.7.16a
module load samtools/1.9
module load htslib bedtools/2.30.0
module load hic-pro/2.11.1
source activate /home/yli11/.conda/envs/captureC
Then submit job:
bsub -q priority -P Genomics -R 'rusage[mem=60000]' -J FIT /home/yli11/Programs/FitHiChIP/FitHiChIP_HiCPro.sh -C hichip.config.txt