CUT & RUN calibration pipeline¶
usage: cut_run_calibration.py [-h] [-j JID] -f FASTQ_TSV -d PEAKCALL_TSV
[--scale_multiplier SCALE_MULTIPLIER]
[--Ecoli_index_file ECOLI_INDEX_FILE]
[-g GENOME] [--genome_fasta GENOME_FASTA]
[-b BLACK_LIST] [-s CHROM_SIZE] [-i INDEX_FILE]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
cut_run_calibration_yli11_2021-06-23)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
TSV file, 3 columns, peak, bam, output, need absolute
path to file (default: None)
-d PEAKCALL_TSV, --peakcall_tsv PEAKCALL_TSV
TSV file, 3 columns, peak, bam, output, need absolute
path to file (default: None)
--scale_multiplier SCALE_MULTIPLIER
--Ecoli_index_file ECOLI_INDEX_FILE
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, mm10, mm9 (default: hg19)
--genome_fasta GENOME_FASTA
genome version: hs, mm (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
-b BLACK_LIST, --black_list BLACK_LIST
Blacklist file (default: /home/yli11/Data/Human/hg19/a
nnotations/hg19.blacklist.bed)
-s CHROM_SIZE, --chrom_size CHROM_SIZE
chrome size (default: /home/yli11/Data/Human/hg19/anno
tations/hg19.chrom.sizes)
-i INDEX_FILE, --index_file INDEX_FILE
chrome size (default: /home/yli11/Data/Human/hg19/inde
x/bwa_16a_index/hg19.fa)
Summary¶
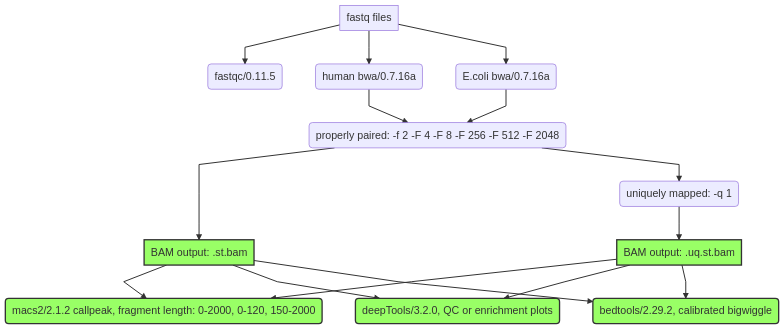
CUN-RUN pipeline with calibrated bw files based on E.coli reads, see https://github.com/Henikoff/Cut-and-Run/blob/master/spike_in_calibration.csh.
Duplicated reads were not removed. Multi-mapped reads can be removed, so two mapped bam files were generated, st.bam is all properly paired reads. .uq.st.bam is all properly paired and uniquely mapped reads.
Bw files, peak files, and motif discovery for top200 peaks were generated for both st.bam and .uq.st.bam files.
Usage¶
Step 0: LOGIN to a compute node
hpcf_interactive
Go to your data directory and type the following.
Step 1: Prepare input files, generate fastq.tsv.
module load python/2.7.13
HemTools cut_run --guess_input
Input fastq files preparation complete! ALL GOOD!
Please check if you like the computer-generated labels in : fastq.tsv
Input peakcall file preparation complete! File name: peakcall.tsv
Note
If you are preparing fastq.tsv and peakcall.tsv yourself, please make sure no space anywhere in the file. Note that the seperator is tab. Spaces in file name will cause errors.
Step 2: Check the computer-generated input list (manually), make sure they are correct.
more fastq.tsv
more peakcall.tsv
Note
a random string will be added to the generated files (e.g., fastq.94c049cbff1f.tsv) if they exist before running step 1.
Step 3: Submit your job.
cut_run_calibration.py -f fastq.tsv -d peakcall.tsv
Sample input format¶
fastq.tsv
This is a tab-seperated-value format file. The 3 columns are: Read 1, Read 2, sample ID.

peakcall.tsv
This is also a tab-seperated-value format file. The 3 columns are: treatment sample ID, control/input sample ID, peakcall ID.

Output¶
See each folder in the {jid} folder for results. For example, calibrated bw files are located in bw_files.
Report bug¶
Once the job is finished, you will be notified by email with some attachments. If no attachment can be found, it might be caused by an error. In such case, please go to the result directory (where the log_files folder is located) and type:
$ HemTools report_bug
Comments¶
code @ github.