Call IDR peaks given bam files from two replicates¶
usage: idr_peaks.py [-h] [-j JID] -r1 R1_INPUT -r2 R2_INPUT
[--merged_input MERGED_INPUT]
[--macs2_addon_parameters MACS2_ADDON_PARAMETERS]
[--half_width HALF_WIDTH] [-g GENOME]
[--macs_genome MACS_GENOME] [-b BLACK_LIST]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
idr_peaks_yli11_2020-05-16)
-r1 R1_INPUT, --R1_input R1_INPUT
TSV file, 2 columns, treatment, control files for
replicate 1 (default: None)
-r2 R2_INPUT, --R2_input R2_INPUT
TSV file, 2 columns, treatment, control files for
replicate 2 (default: None)
--merged_input MERGED_INPUT
Not for end-user anymore (default: None)
--macs2_addon_parameters MACS2_ADDON_PARAMETERS
--half_width HALF_WIDTH
half.width: -1 if using the reported peak width, a
numerical value to truncate the peaks to +- half_width
(default: 200)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, mm10, mm9 (default: hg19)
--macs_genome MACS_GENOME
genome version: hs, mm (default: hs)
-b BLACK_LIST, --black_list BLACK_LIST
Blacklist file (default: /home/yli11/Data/Human/hg19/a
nnotations/hg19.blacklist.bed)
Summary¶
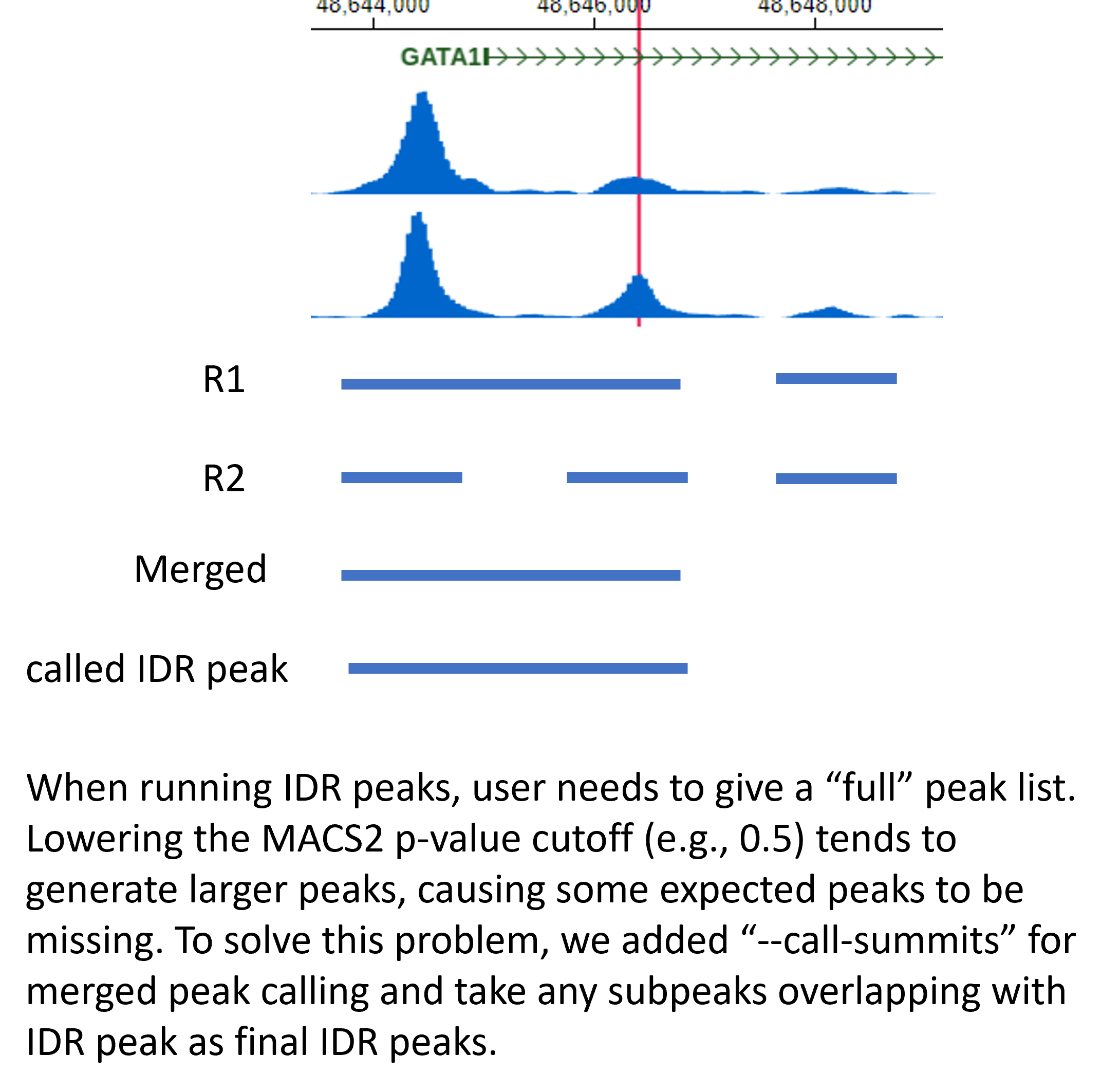
IDR peaks are conserved binding peaks that usually can boost motif enrichment. Note that peaks called from individual replicate can be still useful.
Also note that MACS2 peak calling is bad for broad peaks. So this pipeline is not suitable for broad peaks.
In the output, you will receive two emails. One is the link to the GREAT analysis (i.e., peak annotations). The other one is a notification of job completion.
10/30/2019
Parameters have been updated. Basically MACS2 callpeak uses -p 0.2 cutoff to produce more peaks, then top 500K is used. With these changes, the number of final peaks should increase. We expect the number of IDR peaks (cutoff at 5%) should be around 10K to 40K. One can use --macs2_addon_parameters " -p 0.05" to control the number of called peaks, and these will decrease the number of final peaks.
5/16/2020
There is a large peak width issue. The problem is originated from a relaxed p-value (>0.01) in MACS2 that often makes the peak width larger. The problem is simply solved by truncating the peaks by redefining peak width using the summit and extend +- half.width and then use the new peaks as input to the IDR algorithm.
Note
If you are using peaks based on the MACS peak caller, then use p.value as the ranking measure and also set max peak half-width to 200 bp (since MACS tends to call wider peaks with relaxed p-value thresholds of 1e-2)
ref:
6/16/2021
Update code for atac-seq idr peaks for multiple samples. Note that for chip-seq samples, the old code doesn’t support multiple samples submission.
10/2/2023
Fixed a “bug”.
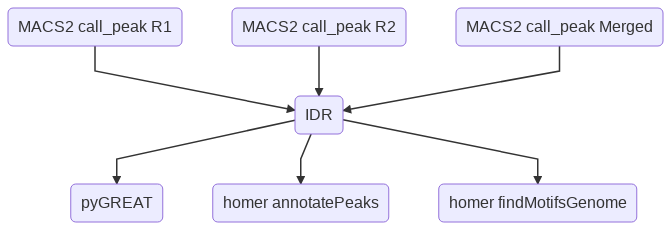
Flowchart¶
Input¶
Please provide the file location to bam files. For single-end data, please use raw bam file (e.g., *.markdup.bam). For paired-end data, please use uniquely mapped de-duplicated bam file (e.g., *.rmdup.uq.bam).
Note
By default, MACS2 will remove duplicated reads. You can specify --keep-dup=all to use all reads.
Note
Currently, this pipeline doesn’t support running multiple samples at the same time. If you have multiple samples, you need to provide the following input files seperately.
R1 Input
This is a two-column tsv file (treatment R1 and contol R1). An example is shown below:
/path_to_file/1047954_Hudep2_CTCF_IP_50bp.markdup.bam /path_to_file/1047955_Hudep2_input_50bp.markdup.bam
R2 Input
This is a two-column tsv file (treatment R2 and contol R2). An example is shown below:
/path_to_file/1047954_Hudep2_CTCF_IP_50bp_R2.markdup.bam /path_to_file/1047955_Hudep2_input_50bp_R2.markdup.bam
Usage¶
Go to your data directory and type the following.
Step 0: Load python version 2.7.13.
module load python/2.7.13
Step 1: Run the program
idr_peaks.py -r1 R1_input -r2 R2_input -g hg19 --macs_genome hs
Note that if you are working on mouse genome, you have to change both -g and --macs_genome options, for example:
idr_peaks.py -r1 R1_input -r2 R2_input -g mm9 --macs_genome mm
For PE-data use:
idr_peaks.py -r1 R1_input -r2 R2_input -g hg19 --macs_genome hs --macs2_addon_parameters " -f BAMPE"
For shorter peak width, pealse add half_width option:
idr_peaks.py -r1 R1_input -r2 R2_input -g hg19 --macs_genome hs --macs2_addon_parameters " -f BAMPE" --half_width 200
ATAC-seq IDR PEAKS¶
usage: idr_peaks_atac.py [-h] [-j JID] -f INPUT_LIST
[--macs2_addon_parameters MACS2_ADDON_PARAMETERS]
[--half_width HALF_WIDTH] [-g GENOME]
[--macs_genome MACS_GENOME] [-b BLACK_LIST]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
idr_peaks_atac_yli11_2021-06-16)
-f INPUT_LIST, --input_list INPUT_LIST
TSV file, 3 columns, Rep1 bam , Rep2 bam, and output
name (default: None)
--macs2_addon_parameters MACS2_ADDON_PARAMETERS
--half_width HALF_WIDTH
half.width: a numerical value to truncate the peaks to
+- half_width (default: 200)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, mm10, mm9 (default: hg19)
--macs_genome MACS_GENOME
genome version: hs, mm (default: hs)
-b BLACK_LIST, --black_list BLACK_LIST
Blacklist file (default: /home/yli11/Data/Human/hg19/a
nnotations/hg19.blacklist.bed)
Input¶
A tsv file containing 3 columns: Rep1 bam , Rep2 bam, and output
Usage¶
module load python/2.7.13
idr_peaks_atac.py -f input.list -g hg19 --macs_genome hs
idr_peaks_atac.py -f input.list -g mm9 --macs_genome mm
Output¶
IDR peaks is shown in idr_peaks.rmblck.bed
You can also find outputs from homer analysis: homer_motifs_result and idr_peaks.annotated.tsv
Ref: https://hbctraining.github.io/Intro-to-ChIPseq/lessons/07_handling-replicates-idr.html
IDR on broad peaks¶
Conclusion: not good mainly because of broad peak calling.
https://github.com/ENCODE-DCC/chip-seq-pipeline2/issues/30
https://groups.google.com/g/idr-discuss/c/_a_GKfw7kwM?pli=1