SHARE-seq data analysis¶
usage: shareseq.py [-h] [-j JID] -f1 SAMPLE_BARCODE -f2 CELL_BARCODE -r1 R1
-r2 R2 [-n NUM_MISMATCH]
[--collision_threshold COLLISION_THRESHOLD]
[--min_reads_per_cell MIN_READS_PER_CELL] [--collision]
[--filter_polyT] [-g GENOME]
[--genome_config GENOME_CONFIG]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
shareseq2_yli11_2021-08-04)
-f1 SAMPLE_BARCODE, --sample_barcode SAMPLE_BARCODE
input config file,tsv: label, sample_barcode, ATAC/RNA
(default: None)
-f2 CELL_BARCODE, --cell_barcode CELL_BARCODE
a list of barcode sequences (default: None)
-r1 R1 input undetermined R1 fastq.gz (default: None)
-r2 R2 input undetermined R2 fastq.gz (default: None)
-n NUM_MISMATCH, --num_mismatch NUM_MISMATCH
number of mismatch allowed (default: 1)
--collision_threshold COLLISION_THRESHOLD
max mapping rate as collision (default: 0.8)
--min_reads_per_cell MIN_READS_PER_CELL
minimal number of reads per cell (default: 100)
--collision map to hybrid genome and calculate collision rate
(default: False)
--filter_polyT polyT reads may not be noise (default: False)
Genome Info:
-g GENOME, --genome GENOME
genome version, must match key in genome config yaml
file (default: hg19)
--genome_config GENOME_CONFIG
genome config file specifing: index file, black list,
chrom size and effectiveGenomeSize (default:
genome.yaml)
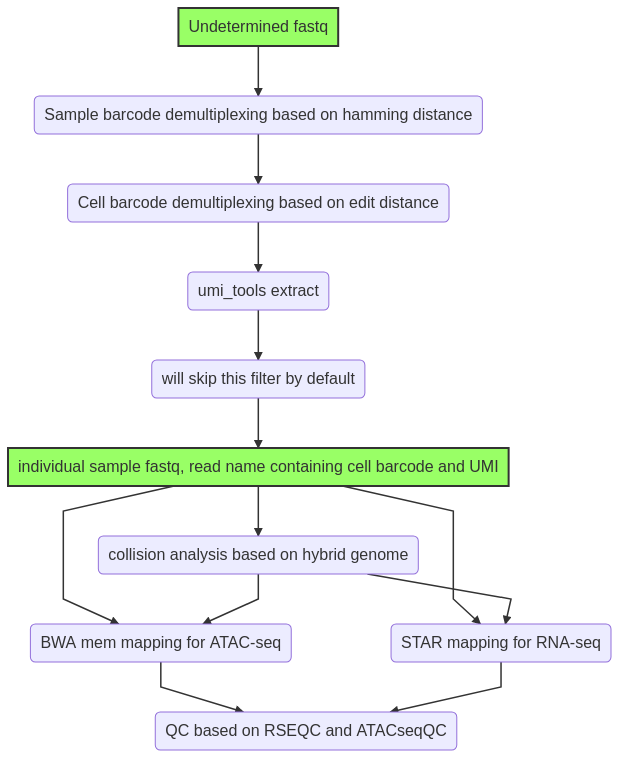
12/7/2023 updates¶
Current ATAC-seq count matrix and fragment file is generated by ArchR. This tool uses a pseudo-bulk (subset of all cells) for peak calling and it only counts reproducible peaks. I found if data quality is not good, this method will miss a lot of candidate peaks.
We will try sinto and signac featurematrix in the future. https://timoast.github.io/sinto/scatac.html#downstream-analysis https://stuartlab.org/signac/articles/peak_calling
Input¶
1. Undetermined_S0_L001_R1_001.fastq.gz and Undetermined_S0_L001_R2_001.fastq.gz¶
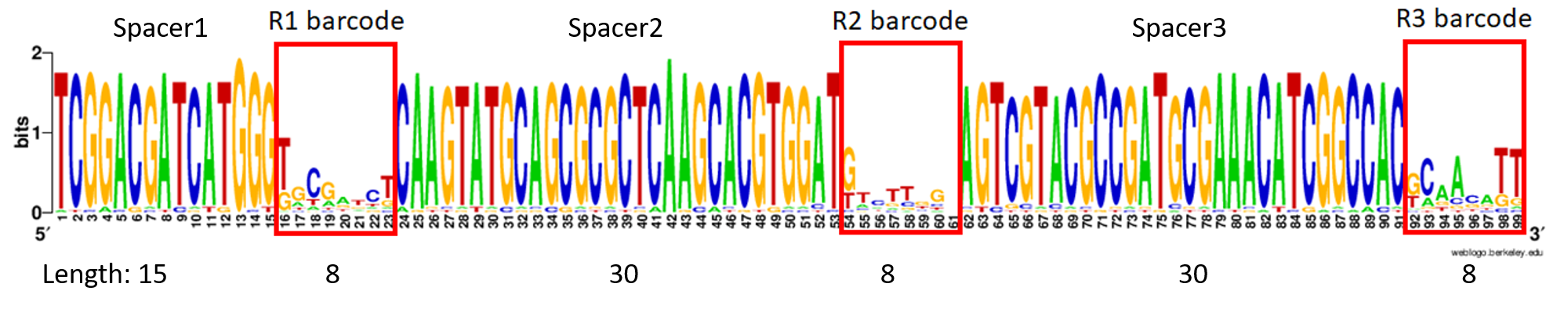
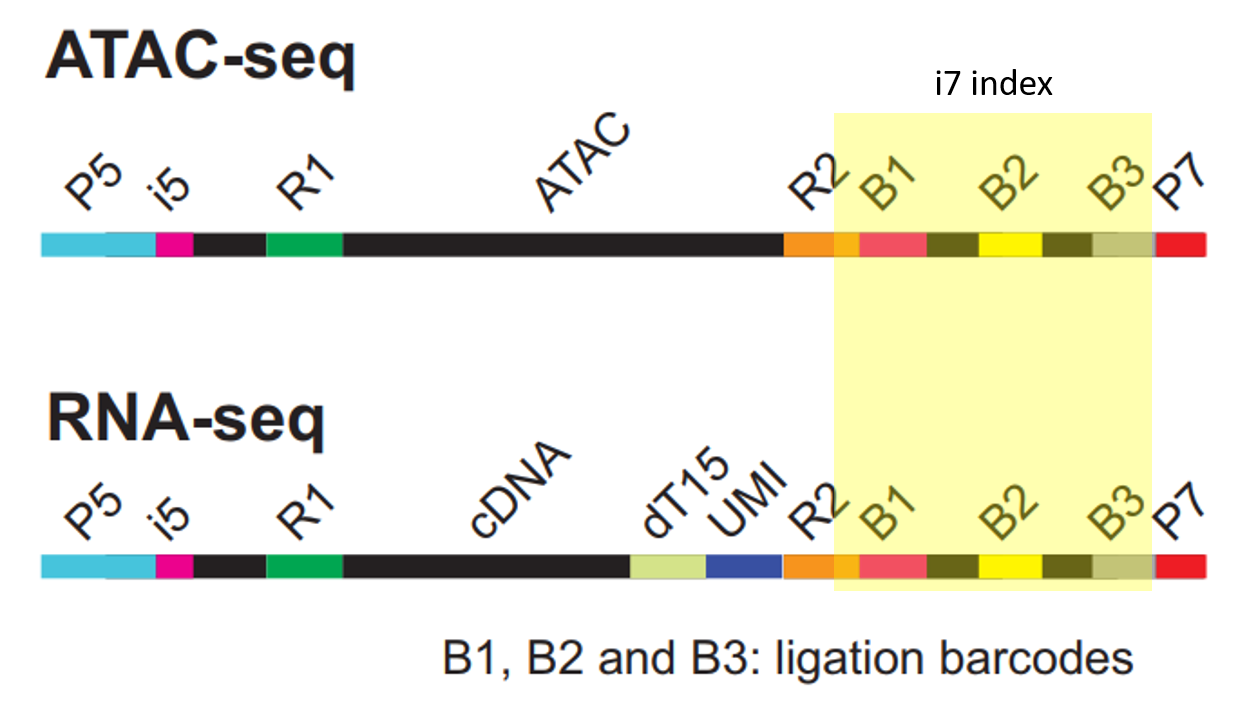
Example read looks like:
@M04990:66:000000000-G83PY:1:1101:15830:1333 1:N:0:TCGGACGATCATGGGGAGCTATTCAAGTATGCAGCGCGCTCAAGCACGTGGATTTTGTTGTAGTCGTACGCCGATGCGAAACATCGGCCACTTTGTTTG+AGAGTAGA
GTATCAACGTTAGTCGTACGCCGATGCGAAACATCGGCCACGAATCTGTA
+
AAAA?33>>AFFFFBAEGAA0EA?AEE0EE//FFAA//EEEEGGAEHBGG
2. input.tsv¶
3-column tsv to specify sample name, sample barcode and sample type (ATAC or RNA)
[yli11@nodecn203 SHARE_seq_pipeline]$ head input.tsv
ATAC_1 TAGATCGC ATAC
RNA_1 TATCCTCT RNA
ATAC_2 CTCTCTAT ATAC
RNA_2 AGAGTAGA RNA
3. barcode.list¶
AACGTGAT
AAACATCG
ATGCCTAA
AGTGGTCA
ACCACTGT
ACATTGGC
CAGATCTG
Usage¶
hpcf_interactive
module load conda3
source activate /home/yli11/.conda/envs/cutadaptenv
# for collision analysis
bsub -q priority -P Genomics -R 'rusage[mem=60000]' -J SHARE python /home/yli11/Tools/SHARE_seq_pipeline/shareseq.py -f1 input.tsv -f2 barcode1.list -r1 Undetermined_S0_L001_R1_001.fastq.gz -r2 Undetermined_S0_L001_R2_001.fastq.gz --collision -n 1 --min_reads_per_cell 10
# for regular share-seq analysis
bsub -q priority -P Genomics -R 'rusage[mem=60000]' -J SHARE python /home/yli11/Tools/SHARE_seq_pipeline/shareseq.py -f1 input.tsv -f2 barcode1.list -r1 Undetermined_S0_L001_R1_001.fastq.gz -r2 Undetermined_S0_L001_R2_001.fastq.gz -n 1 --min_reads_per_cell 10
New case: map to gRNA vector¶
bsub -q priority -P Genomics -R 'rusage[mem=60000]' -J SHARE python /home/yli11/Tools/SHARE_seq_pipeline/shareseq.py -f1 input.tsv -f2 barcode1.list -r1 Undetermined_S0_R1_001.fastq.gz -r2 Undetermined_S0_R2_001.fastq.gz -n 1 --min_reads_per_cell 10 --vector /home/yli11/Tools/SHARE_seq_pipeline/vector.fa
Output¶
Look for collision plot.
CROP gRNA¶
fastq.tsv, fastq, label, gRNA library file
CROP_1200_C3.fastq.gz C1200_gRNA C1200_gRNA_library.csv
CROP_6991_C4.fastq.gz C6991_gRNA C6991_gRNA.csv
fastq file looks like:
@M04990:171:000000000-GCTR9:1:1101:17215:1578 1:N:0:TCGGACGATCATGGGGTGTGTCGCATGTATGCAGTGCGCTCAAGCACGTGGATTTGCGTACTGTCGTTCGTTGATGTGTTTTATCTGTTATTGTATGTT
GCAGAGTCGGCTTTATATATCTTGTGGAAAGGACGAAACACCGACATGTAGATTTTAGAGCTAGAAATAGCAAGATAAAAAAAAAAA
+
1A111>BF11AAGGFGFGFFFGHFGC0111000BA00EA0FEC///AEGB2HHHHEECCF1B@FBF1B@FFG1F0BFBG1F/>>>/<
gRNA library csv need to have header:
sgRNA,seq,gene
name,AXXXXAAGAGGCAACTG,gene_name
run_lsf.py -f fastq.tsv -p CROP_gRNA