Call motif footprint from bigwiggle files¶
usage: motif_footprint_from_bw.py [-h] [-j JID] -f INPUT_LIST [-m MOTIF_FILE]
[-c FIMO_CUTOFF] [-g GENOME]
[--genome_fasta GENOME_FASTA]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
motif_footprint_from_bw_yli11_2020-07-12)
-f INPUT_LIST, --input_list INPUT_LIST
tsv 3 columns, bed,bw,outputname (default: None)
-m MOTIF_FILE, --motif_file MOTIF_FILE
motif file, use absolute path (default: /home/yli11/Da
ta/Motif_database/Human/homer_jaspar.meme)
-c FIMO_CUTOFF, --fimo_cutoff FIMO_CUTOFF
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19, mm10, mm9 (default: hg19)
--genome_fasta GENOME_FASTA
genome version: hs, mm (default:
/home/yli11/Data/Human/hg19/fasta/hg19.fa)
Summary¶
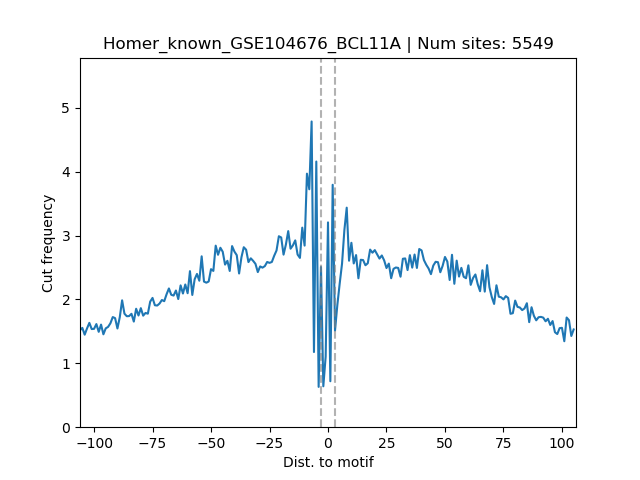
This pipeline is designed as the next step after ATAC-seq footprint pipeline. Given motif pwm file, we first perform motif scanning on the given peak file, we then extract Tn5 cutting frequency from the bw file. A valid motif footprint should be a U-shape (i.e., mean frequency inside the motif is smaller than the flanking regions).
Input¶
The input file is a tsv format containing 3 columns: bed file, bw file, output-prefix.
For bed file, either relative path or absolute path is OK.
For bw file, it needs absolute path.
H2.narrowPeak /path/to/H2.bw Hudep2_footprints
H1.narrowPeak /path/to/H1.bw Hudep1_footprints
Usage¶
module load python/2.7.13
motif_footprint_from_bw.py -f input.list
For mouse genome
module load python/2.7.13
motif_footprint_from_bw.py -f input.list -g mm9 -m /home/yli11/Data/Motif_database/Mouse/mouse_TF.meme
Output¶
Each line will have its own result dir in the jobID folder.
Individual motif footprint result is shown in jobID/output_name/FIMO_motif_mapping/ directory.
In each motif folder, you can find:
the exact mapped location:
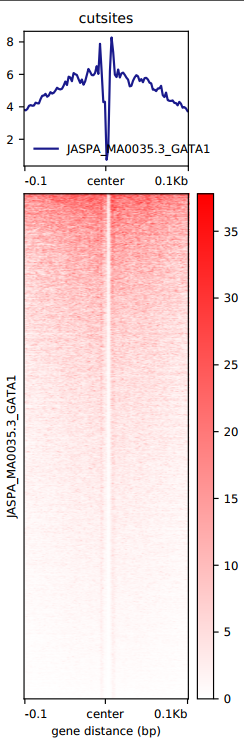
fimo.cuts.freq.txt.filtered_fimo.bedthe average footprint signal plot:
motif_name.footprint.png.
If there’s a motif that you are interested in, there’s a script for you to generate a footprint heatmap plot: signal_plot.sh, which you just need to run bash signal_plot.sh.
“NFIX”,”PU.1”,”CTCF”,”GATA1”,”ZBTB7A” will be automatically generated since they are used very often in our projects.