Variant identification on RNA-seq data¶
Note
Need more than 25M mapped reads for variant calling. Currently, this pipeline only work on hg19.
usage: rna_seq_variant_call.py [-h] [-j JID] [--pipeline_type PIPELINE_TYPE]
[-d DEPTH_FILTER]
(-f FASTQ_TSV | --guess_input) [-g GENOME]
[--STAR_index STAR_INDEX]
optional arguments:
-h, --help show this help message and exit
-j JID, --jid JID enter a job ID, which is used to make a new directory.
Every output will be moved into this folder. (default:
rna_seq_variant_call_yli11_2019-07-09)
--pipeline_type PIPELINE_TYPE
Not for end-user. (default: rna_seq_variant_call)
-d DEPTH_FILTER, --depth_filter DEPTH_FILTER
filter variants by read depth (default: 10)
-f FASTQ_TSV, --fastq_tsv FASTQ_TSV
tab delimited 3 columns (tsv file): Read 1 fastq, Read
2 fastq, sample ID (default: None)
--guess_input Let the program generate the input files for you.
(default: False)
Genome Info:
-g GENOME, --genome GENOME
genome version: hg19. Only working for hg19 (default:
hg19)
--STAR_index STAR_INDEX
genome version: hg19. Only working for hg19 (default:
/research/dept/hem/common/sequencing/chenggrp/pipeline
s/hg19/hg19_star_253a_index/)
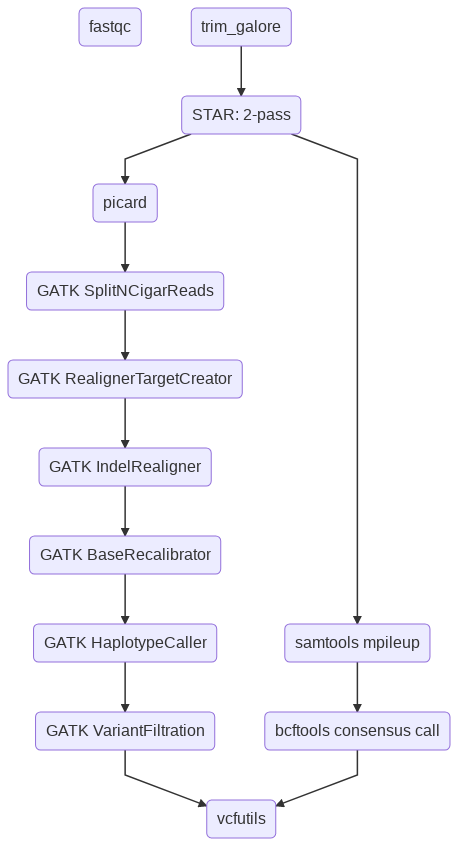
Summary¶
Perform RNA-seq variant calling using STAR alignment followed by the GATK pipeline and the samtools/bcftools pipeline.
The GATK pipeline is optimized for RNA-seq data.
The samtools/bcftools pipeline is a general-purpose variant calling pipeline for NGS data.
Flowchart¶
Usage¶
Go to your data directory and type the following.
Step 0: Load python version 2.7.13.
module load python/2.7.13
Step 1: Prepare input files, generate fastq.tsv.
rna_seq_variant_call.py --guess_input
Input fastq files preparation complete! ALL GOOD!
Please check if you like the computer-generated labels in : fastq.tsv
Note
If you are preparing fastq.tsv yourself, please make sure no space anywhere in the file. Note that the seperator is tab. Spaces in file name will cause errors.
Step 2: Check the computer-generated input list (manually), make sure they are correct.
$ less fastq.tsv
Note
a random string will be added to the generated files (e.g., fastq.94c049cbff1f.tsv) if they exist before running step 1.
Step 3: Submit your job.
rna_seq_variant_call.py -f fastq.tsv
Read depth
By default, variants that have less than 10 read depth will be filtered. You can increase the threshold using -d option.
Sample input format¶
fastq.tsv
This is a tab-seperated-value format file. The 3 columns are: Read 1, Read 2, sample ID.

Output¶
Final filtered VCF files are located in the final_results folder.
{{output_name}}.GTAK.bcftools.final.vcf is the result from the GATK pipeline.
{{output_name}}.samtools.final.vcf is the result from the samtools/bcftools pipeline.
Variant calling comparing to WT¶
With --WT option, you can filter out low-confident variant using a WT RNA-seq data. Same pipeline was used to generate the figure below in our ABE paper.
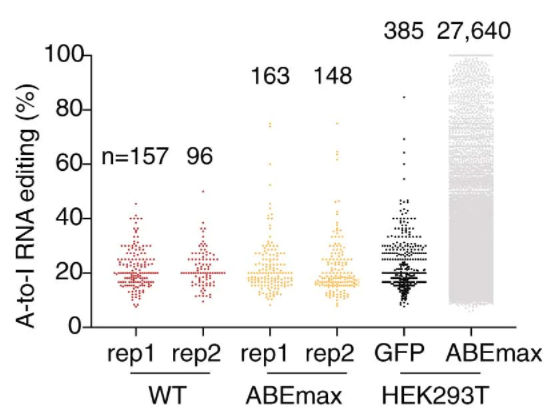
rna_seq_variant_call.py -f fastq.tsv -WT Hudep2_WT
Output is in compare_result/*all_passed_variants.csv.
Reference¶
https://software.broadinstitute.org/gatk/documentation/article.php?id=3891