Genome-wide CRISPR Screening¶
usage: HemTools crispr_seq [-h] [-j JID] [--short] [--debug] [--count_only]
[--paired] [--control_gRNAs CONTROL_GRNAS]
[-d DESIGN_MATRIX] --gRNA_library GRNA_LIBRARY
[--bed BED] [-g GENOME]
Named Arguments¶
- -j, --jid
enter a job ID, which is used to make a new directory. Every output will be moved into this folder.
Default:
'{{subcmd}}_docs_2026-04-30'- --short
Force to use the short queue.
Default:
False- --debug
Not for end-user.
Default:
False- --count_only
Only perform Mageck count
Default:
False- --paired
Mageck RRA paired test, this is not available for mageck MLE
Default:
False
Input files¶
- --control_gRNAs
a list of control gRNAs. If provided, normalization will be performed based on these controls, instead of median normalization.
- -d, --design_matrix
tab delimited 3 columns (tsv file): treatment sample ID, control sample ID, peakcall ID
- --gRNA_library
mageck format
Genome Info¶
- --bed
Genomic coordinates for gRNAs (Format: chr, start, end, name). If provided, raw counts, logFC, logFDR will be uploaded to protein paint for visualization.
- -g, --genome
genome version: hg19, hg38
Default:
'hg19'
Input file¶
INPUT 0. fastq files.
No options to input a list of fastq files. All *.fastq.gz files in the current directory will be used.
Inf you don’t want to use all fastq files, please create a new directory, cd to that directory, and create soft links for the files you need. A soft link is similar to file shortcut used in Windows.
The command to create a soft link is:
$ ln -s [original filename] [link name]
INPUT 1. Design matrix (-d option, required).
This file specifies a list of pairwise comparisons. Each comparison has a comparison name, a list of control group files, and a list of treatment group files. The format is a 3-column tsv file. An example file is shown below.
APC control CRM-APC-Negative-rep1_S10_L001_R1_001.fastq.gz,CRM-APC-Negative-rep2_S11_L001_R1_001.fastq.gz
APC treatment CRM-APC-positive-rep1_S4_L001_R1_001.fastq.gz,CRM-APC-positive-rep2_S5_L001_R1_001.fastq.gz
GFP control CRM-GFP-Negative-rep1_S7_L001_R1_001.fastq.gz,CRM-GFP-Negative-rep2_S8_L001_R1_001.fastq.gz
GFP treatment CRM-GFP-Positive-rep1_S1_L001_R1_001.fastq.gz,CRM-GFP-Positive-rep2_S2_L001_R1_001.fastq.gz
APC_neg_vs_plasmid control plasmid.fastq.gz
APC_neg_vs_plasmid treatment CRM-APC-Negative-rep1_S10_L001_R1_001.fastq.gz,CRM-APC-Negative-rep2_S11_L001_R1_001.fastq.gz
APC_pos_vs_plasmid control plasmid.fastq.gz
APC_pos_vs_plasmid treatment CRM-APC-positive-rep1_S4_L001_R1_001.fastq.gz,CRM-APC-positive-rep2_S5_L001_R1_001.fastq.gz
Note
Comparison names (column 1) should be unique. control & treatment (column 2) are keywords, misspelling can cause error.
INPUT 2. gRNA library csv file (–gRNA_library option, required).
This file specifies your gRNA library. It is a csv file where the columns are sgRNA, sgRNA sequence, and the targeted gene. This file must end with .csv. An example file is shown below.
sgRNA,Sequence,Gene
chr11:4167629-AAATTTCCTCAGCAGATTAC,AAATTTCCTCAGCAGATTAC,Gene1
Please_no_space_anywhere,ACAAGCAACAGTTGACCAAC,Gene1
could_be_anything,ACATGAGACTGGAAACCGCC,positive_control
INPUT 3. control gRNA list (–control_gRNAs option, optional).
The file specifies a list of control gRNA names. The names should match to the gRNA library file. Each line is a control gRNA name.
Tip
control gRNAs are optional. If provided, normalization will be performed based on these controls, instead of median normalization.
INPUT 4. Genomic coordinate bed file of gRNA (–bed option, optional).
Genomic coordinates for gRNAs (at least 4 columns: format: chr, start, end, name). If provided, raw counts, logFC, logFDR will be uploaded to protein paint for visualization. An example file is shown below. The last column name should be either gRNA id (i.e., the 1st column in the gRNA lib file.)
chr11 4167629 4167649 sgRNA1_id
Tip
gRNAs locations are optional. If provided, raw counts, logFC, logFDR will be uploaded to protein paint for visualization. gRNA strand info is not required.
Usage¶
Go to your data directory and type the following.
Step 0: Load python version 2.7.13.
$ module load python/2.7.13
**Step 1: Prepare the input files, see the format above. **
Note
Please make sure there is no space anywhere in file name, sgRNA names, and gene names.
Step 2: Submit your job.
$ HemTools crispr_seq -d design_matrix.tsv --gRNA_library my_gRNAs.csv --control_gRNAs my_controls.list
OR:
$ HemTools crispr_seq -d design_matrix.tsv --gRNA_library my_gRNAs.csv --control_gRNAs my_controls.list --bed my_gRNAs.bed
OR you can perform MaGeCK RRA paired test by add --paired option:
Note
Paired test is only available for MaGeCK RRA method, not available for the MLE method.
Note
In paired mode, the number of control samples must be the same as the number of treatment samples.
$ HemTools crispr_seq -d design_matrix.tsv --gRNA_library my_gRNAs.csv --control_gRNAs my_controls.list --bed my_gRNAs.bed --paired
Increase mapping rate¶
If you have different length of gRNAs in your library, automatic determination may decrease the gRNA mapping rate. In these cases, you may want to fix the gRNA search positions, such as --trim_5 40,41,42,43,44,45,46
trim position depends on your library preparation. In Cheng lab, it is mostly at 43.
The complete command is:
$ HemTools crispr_seq -d design_matrix.tsv --gRNA_library my_gRNAs.csv --control_gRNAs my_controls.list --bed my_gRNAs.bed --trim_5 40,41,42,43,44,45,46
Output files¶
QC¶
In the email attachment, you can find Mageck count report, example shown below.
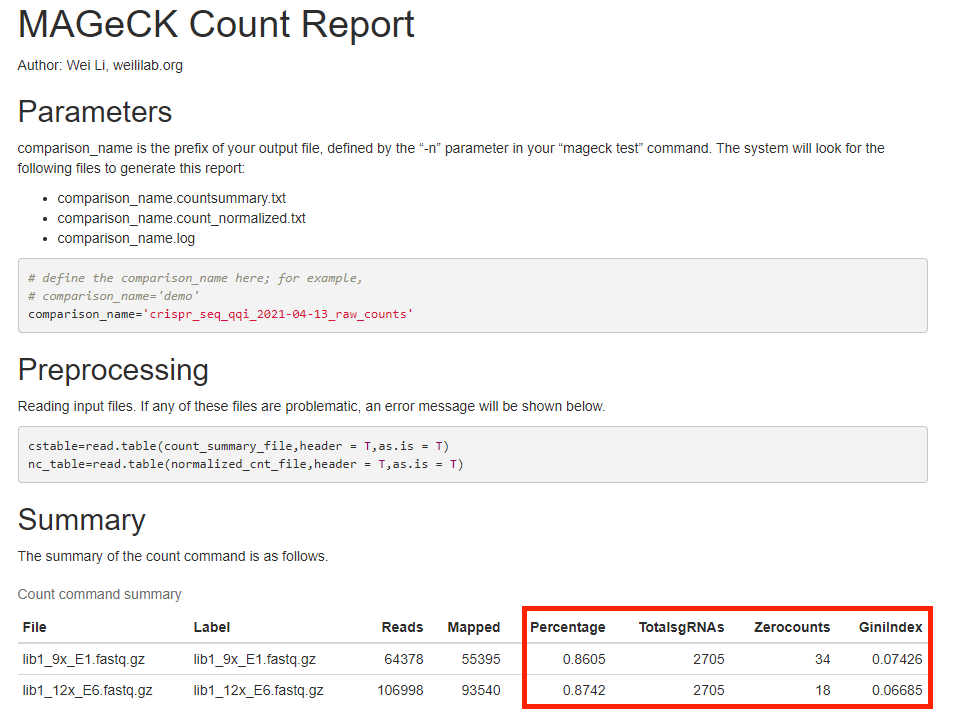
Here you can check mapping rate, number of sgRNAs with zero count, and gini index for eveness (<0.2 is good).
Report bug¶
Once the job is finished, you will be notified by email with some attachments. If no attachment can be found, it might be caused by an error. In such case, please go to the result directory (where the log_files folder is located) and type:
$ HemTools report_bug
TODO¶
HPC doesn’t have the latest version of Mageck. A request has been submitted.
For the insulator project¶
add --kallisto option to use kallisto to map R1 read to 250bp fasta library and count reads, generates Mageck format count table so that we can use Mageck to gengerate visualizations and comparisons.
Comments¶
code @ github.