HemTools Tutorial 4-18-2019¶
Step 0: set up RSA authentication with HPC
Create a test run folder¶
cd ~
mkdir my_test_run
cd my_test_run
Installation¶
/research/dept/hem/common/sequencing/chenggrp/pipelines/bin/add_env.sh
Type HemTools -h to see a list of available pipelines.
HemTools: performs NGS pipelines and other common analyses. Contact:
Yichao.Li@stjude.org or Yong.Cheng@stjude.org
positional arguments:
{cut_run,chip_seq_pair,chip_seq_single,atac_seq,report_bug,rna_seq,my_dir,volcano_plot,crispr_seq}
Available APIs in HemTools
cut_run CUT & RUN pipeline
chip_seq_pair Paired-end ChIP-seq pipeline
chip_seq_single Single-end ChIP-seq pipeline
atac_seq ATAC-seq pipeline
report_bug Email the log files to the developer.
rna_seq RNA-seq pipeline
my_dir CD, search, and list my dirs
volcano_plot Data visualization: Volcano plot
crispr_seq Genome-wide CRISPR Screening pipeline
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit
Workflow¶
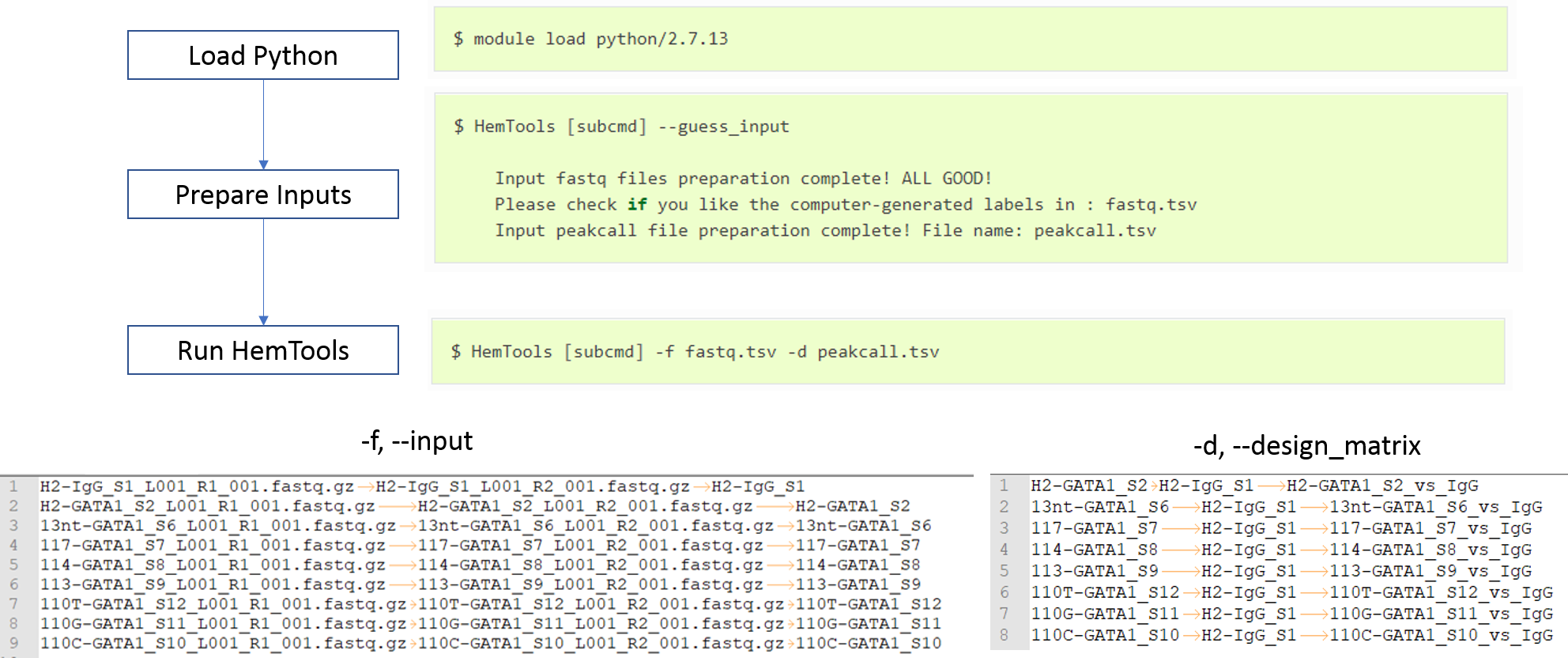
Note
Since the upgrade from Red Hat 6 to 7, the --short option is not working anymore. Just don’t add this option to your program is OK.
ATAC-seq example¶
Copy data
mkdir atac_seq
cd atac_seq
ln -s /research/dept/hem/common/sequencing/chenggrp/pipelines/example_data/atac_seq/*.gz .
Run HemTools
module load python/2.7.13
HemTools atac_seq --guess_input
HemTools atac_seq -f fastq.tsv --short
Single-end ChIP-seq example¶
Copy data
cd ..
mkdir chip_seq_single
cd chip_seq_single
ln -s /research/dept/hem/common/sequencing/chenggrp/pipelines/example_data/chip_seq_single/*.gz .
Run HemTools
module load python/2.7.13
HemTools chip_seq_single --guess_input
HemTools chip_seq_single -f fastq.tsv -d peakcall.tsv --short
Paired-end ChIP-seq example¶
Copy data
cd ..
mkdir chip_seq_pair
cd chip_seq_pair
ln -s /research/dept/hem/common/sequencing/chenggrp/pipelines/example_data/chip_seq_pair/*.gz .
Run HemTools
module load python/2.7.13
HemTools chip_seq_pair --guess_input
HemTools chip_seq_pair -f fastq.tsv -d peakcall.tsv --short
CUT & RUN example¶
Copy data
cd ..
mkdir cut_run
cd cut_run
ln -s /research/dept/hem/common/sequencing/chenggrp/pipelines/example_data/cut_run/*.gz .
Run HemTools
module load python/2.7.13
HemTools cut_run --guess_input
HemTools cut_run -f fastq.tsv -d peakcall.tsv --short
Report bug¶
Once the job is finished, you will be notified by email with some attachments. If no attachment can be found, it might be caused by an error. In such case, please go to the result directory (where the log_files folder is located) and type:
cd ..
cd atac_seq
cd [YOUR_JOB_ID]
HemTools report_bug
Questionnaire¶
Please take a minute to complete this HemTools Tutorial questionnaire
Comments¶
code @ github.